Net zo Engelmann-syndroom wordt een zeldzame vorm van osteosclerose genoemd. Het wordt geassocieerd met een vermindering van de veerkracht van de botten en andere symptomen en kan met verschillende medicijnen worden behandeld.
Wat is het Engelmann-syndroom?
© sveta - stock.adobe.com
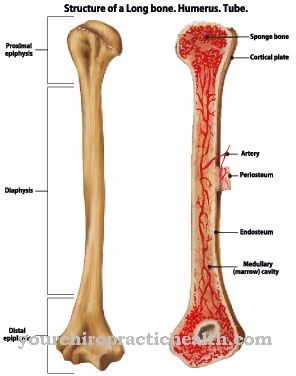
Het Engelmann-syndroom is een vorm van osteosclerose die werd ontdekt door de Duitse fysioloog Theodor Wilhelm Engelmann. Het wordt gekenmerkt door een aantal symptomen zoals een toenemende botverharding en een gelijktijdige vermindering van de botweerstand.
Omdat de ziekte wordt veroorzaakt door een genmutatie, is preventie in de klassieke zin niet mogelijk. Na een uitgebreide diagnose, inclusief diverse röntgenonderzoeken, kunnen getroffenen echter met verschillende medicijnen worden behandeld. Als dit vroeg gebeurt, kan een positief verloop van de ziekte worden verwacht.
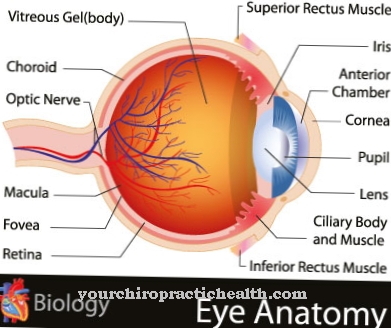
Als de behandeling echter wordt uitgesteld, verspreiden de symptomen zich naar de schedelbasis, wat kan leiden tot blindheid, aangezichtsverlamming en doofheid. Aan de andere kant kan de vernauwing van de zenuwkanalen in het ergste geval de hersenen beschadigen.
oorzaken
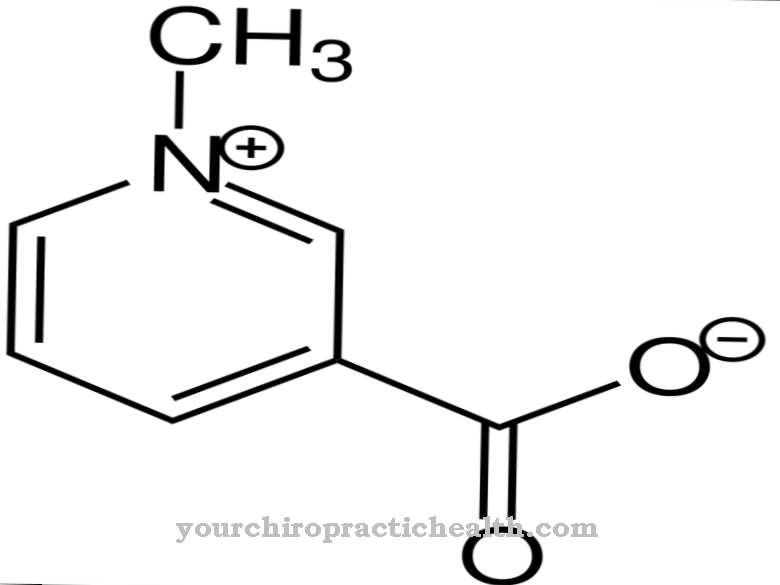
Het Engelmann-syndroom wordt veroorzaakt door een mutatie op chromosoom 19. Meer bepaald door een verandering in de genlocus 19q13.1-13.3, die de zogenaamde transformerende groeifactor beschadigt. Ook hier is de beta-1-keten van het molecuul defect. Omdat de TFG verantwoordelijk is voor de vorming van de botten, ontstaan er afwijkingen in de botten en andere aandoeningen bij een genetische fout.
De symptomen kunnen sterk verschillen van drager tot drager, daarom is een uitgebreide diagnose des te belangrijker. De genmutatie wordt overgeërfd als een autosomaal dominante eigenschap, maar er zijn ook sporadische gevallen. Het is dus mogelijk dat de mutatie een generatie overslaat, waarbij de mens als drager blijft dienen en het defecte gen erven.
Uw medicatie vindt u hier
➔ Geneesmiddelen tegen pijnSymptomen, kwalen en tekenen
Mensen met het Engelmann-syndroom kampen al in de vroege kinderjaren met ernstige symptomen. Kort na de geboorte ontwikkelen zich sclerosen en hyperostosen op de lange botten. Aanvankelijk worden alleen de onderbenen aangetast voordat de ziekte zich naar andere botten verspreidt en, indien onbehandeld, uiteindelijk de gehele botstructuur aantast.
Omdat de benige zenuwkanalen ook in het latere verloop van de ziekte worden aangetast, treedt craniale zenuwverlamming op en dit leidt weer tot verdere symptomen en secundaire ziekten. Visuele beperking en doofheid kunnen optreden, evenals totale blindheid en aangezichtsverlamming. De intelligentie blijft hierdoor onaangetast, maar vooral bij kinderen treden hoofdpijn, vermoeidheidssymptomen en concentratiestoornissen op.
Bovendien begint de puberteit vaak laat. Botpijn komt ook voor, er zijn problemen met lopen en de getroffenen lijden aan hypotensie van de ledemaatspieren. Daarnaast zijn er inherente aandoeningen van de spieren, ook wel myopathie genoemd.
Dit leidt vaak tot bloedarmoede en leukopenie en, afhankelijk van het ziektebeeld, tot andere symptomen. Het syndroom van Raynaud, een vaatziekte die herkenbaar is aan de blauwe kleur van de vingers en een bleke huid, komt bijzonder vaak voor.
Diagnose en verloop
De diagnose van het Engelmann-syndroom wordt gesteld door middel van röntgenonderzoek. Hierdoor kunnen typische symptomen zoals vergrote medullaire holtes en een ongebruikelijke verdikking van de corticale laag op de aangetaste lange botten worden vastgesteld. Er worden ook verschillende tests uitgevoerd om andere osteosclerose uit te sluiten, zoals craniodiaphyseale dysplasie.
Vanwege de grote variabiliteit van de symptomen en hun ernst lukt dit echter niet altijd, daarom is een aanvullende medische anamnese nodig. Dit richt zich op gelijkaardige ziekten in de familie van de getroffen persoon, waarbij ook rekening wordt gehouden met het eerste optreden van de symptomen en andere bijzondere kenmerken. Een ziektedagboek kan een nuttige aanvulling zijn op de anamnese.
Verder wordt in een gesprek tussen de arts en de patiënt verduidelijkt hoe hevig de pijn is, waar deze optreedt en of de genoemde verlammingsverschijnselen al zijn opgetreden. Dit maakt het mogelijk om enerzijds vast te stellen of het het syndroom van Engelmann betreft en anderzijds om het stadium van de ziekte te verduidelijken. Het verloop van de ziekte wordt over het algemeen vrij positief beoordeeld.
Met vroege behandeling kunnen de klinische en radiologische afwijkingen uitgebreid worden behandeld, waarbij secundaire symptomen zoals een kleine gestalte niet ongewoon zijn. Als de ziekte niet wordt behandeld, zullen er later andere botten ontstaan en zal uiteindelijk de schedelbasis worden aangetast. Als hier zenuwen bekneld raken, zijn neurologische gebreken en andere, soms fatale symptomen het gevolg.
Complicaties
In de regel is de veerkracht van de botten verminderd bij het Engelmann-syndroom. Zelfs kleine ongevallen of stoten kunnen tot breuken leiden. De patiënt is beperkt in zijn dagelijkse leven en kan geen lichamelijk zwaar werk verrichten.
De meeste patiënten ervaren de eerste complicaties direct na de geboorte. Sclerose vormt zich in het proces. De ziekte verspreidt zich in de loop van het leven naar andere botten en uiteindelijk naar het hele lichaam. Het Engelmann-syndroom heeft ook een negatief effect op het gezichtsvermogen en het gehoor. Volledige doofheid kan het gevolg zijn.
Blindheid komt meestal niet voor. Er zijn geen geestelijke beperkingen bij de patiënt, zodat denken en handelen op de gebruikelijke manier mogelijk is. Door de pijn in de botten ervaren veel mensen ook pijn tijdens het lopen. Dit leidt tot een onnatuurlijke manier van lopen die andere mensen bizar kunnen vinden.
Vooral kinderen en jongeren worden hierdoor gepest. Behandeling is mogelijk door steroïden toe te voegen. Vaak kunnen echter niet alle symptomen van het Engelmann-syndroom worden bestreden. Naast medicamenteuze behandeling kan fysiotherapie ook nuttig en ondersteunend zijn.
Wanneer moet je naar de dokter gaan?
Kinderen die aan het Engelmann-syndroom lijden, moeten vanaf jonge leeftijd medische behandeling krijgen. Meestal wordt de ziekte onmiddellijk na de geboorte gediagnosticeerd en wordt de therapie onmiddellijk gestart. Verdere doktersbezoeken zijn vereist als zich complicaties voordoen of ongebruikelijke symptomen en klachten optreden.
In het geval van plotselinge hoofdpijn, concentratiestoornissen of vermoeidheidsverschijnselen, moet u beslist uw kinderarts of huisarts raadplegen.
Als de typisch voorkomende sclerosen en hyperostosen bewegingsbeperkingen veroorzaken, kunt u het beste een orthopedisch chirurg of chiropractor raadplegen. Bij de eerste tekenen dat de ziekte zich heeft verspreid naar andere botten, moet het kind naar een gespecialiseerde kliniek worden gebracht.
Onmiddellijke medische opheldering is ook vereist bij ernstige botpijn, problemen met lopen en secundaire ziekten zoals bloedarmoede of leukopenie. Als er tekenen zijn van het syndroom van Raynaud, moet een specialist in vaatziekten worden ingeschakeld. Aangezien de getroffen kinderen en hun ouders vaak ook psychisch lijden aan het Engelmann-syndroom, moet therapeutisch advies worden ingewonnen.
Artsen & therapeuten bij u in de buurt
Behandeling en therapie
Het Engelmann-syndroom wordt behandeld met corticosteroïden. Deze leiden tot normalisatie van de verschillende afwijkingen, maar veroorzaken ook een klein postuur en andere bijwerkingen. Er is ook een risico op de ziekte van Cushing. Dit syndroom wordt veroorzaakt door een verhoogd gebruik van bepaalde medicijnen en leidt tot spierzwakte, zwaarlijvigheid van de romp en diverse andere symptomen.
Er zijn geen andere behandelingsopties bekend naast de toediening van cortison. Het is echter mogelijk om bestaande botschade chirurgisch te behandelen. Fysiotherapie kan ook bijdragen aan herstel, maar onder normale omstandigheden kan bestaande botschade niet volledig ongedaan worden gemaakt.
Outlook & prognose
Bij het Engelmann-syndroom is de prognose niet bijzonder goed vanwege de progressieve ziekte. De vooruitzichten zijn iets beter met de zwakkere variant van het Engelmann-syndroom, het zogenaamde ribbelsyndroom.
De gevolgen van de opgetreden osteosclerose wegen zwaar op de getroffenen. Gegeneraliseerde bothypertrofie resulteert in toenemende botverharding. Dit leidt op zijn beurt tot een gebrek aan flexibiliteit en veerkracht in de botten. De genetische gevolgen van de ziekte kunnen niet worden herzien. De getroffenen moeten leren leven met ernstige pijn, beperkte mobiliteit en spierzwakte en algemene vermoeidheid.
De vooruitzichten zijn beter als de getroffen persoon drager is van het gemuteerde gen maar niet lijdt aan het Engelmann-syndroom. Hij wordt dan echter de drager van het genetisch defect aan zijn nageslacht. Aangezien de gevolgen van de ziekte zich manifesteren in de kindertijd, is er nauwelijks iets dat kan worden gedaan om de progressie van de ziekte tegen te gaan. De veel voorkomende bijkomende vaatziekten maken het Engelmann-syndroom moeilijker.
De prognose wordt verbeterd door vroege toediening van corticosteroïden. Dit kan echter de prijs van een klein postuur kosten. Er kan ook een hypofysetumor (de ziekte van Cushing) ontstaan. De voordelen en risico's van dergelijke symptomatische behandelingen moeten daarom zorgvuldig worden afgewogen.
Aangezien de ziekte ondanks alle maatregelen voortschrijdt, zijn de vooruitzichten voor de getroffenen niet al te rooskleurig. De geneeskunde heeft momenteel nauwelijks middelen om de symptomen van het Engelmann-syndroom te verlichten.
Uw medicatie vindt u hier
➔ Geneesmiddelen tegen pijnpreventie
Het Engelmann-syndroom kan nog niet worden voorkomen omdat het een aandoening van de genen is. De enige manier om dit te voorkomen, is door een mutatie bij de foetus te diagnosticeren. Passende maatregelen zoals het toedienen van medicatie kunnen dan in een vroeg stadium worden ingezet, waardoor langdurige schade tot een minimum wordt beperkt.
Vanaf dat moment kunnen getroffenen de begeleidende symptomen in ieder geval grotendeels voorkomen door middel van fysiotherapie, sport en het gebruik van genoemde medicijnen.
Nazorg
Nazorgmaatregelen zijn zeer beperkt bij het Engelmann-syndroom. Aangezien een volledige genezing niet kan worden bereikt, is de focus van de behandeling de vroege diagnose en de vroege start van de behandeling. Alleen zo kunnen verdere complicaties en klachten worden voorkomen.
In de meeste gevallen zijn patiënten afhankelijk van medicatie en steroïden. Ernstige complicaties komen zelden voor, maar de getroffenen moeten er altijd voor zorgen dat de medicatie correct en regelmatig wordt ingenomen. Ook de instructies van de arts dienen in acht te worden genomen en bij twijfel of andere onzekerheden dient ook een arts te worden geraadpleegd.
In veel gevallen zijn de getroffenen ook afhankelijk van revalidatiemaatregelen zoals fysiotherapie. Dit moet regelmatig gebeuren. Veel oefeningen uit de fysiotherapie of fysiotherapie kunt u ook bij u thuis uitvoeren. Dit kan het genezingsproces voor de getroffen persoon versnellen.
De patiënten zijn meestal ook afhankelijk van regelmatig onderzoek door een arts. Dit is de enige manier om verdere complicaties te voorkomen. Het is niet universeel te voorspellen of het Engelmann-syndroom zal leiden tot een verminderde levensverwachting. In sommige gevallen kan contact met andere patiënten met het syndroom ook nuttig zijn.
U kunt dat zelf doen
Om ervoor te zorgen dat de patiënt zijn bewegingsvrijheid zo lang mogelijk behoudt, moeten gerichte bewegingen en training worden uitgevoerd. Deze kunnen meerdere keren per dag zelfstandig worden gebruikt. Vaak zijn kleine oefeneenheden binnen de dagelijkse routines en processen voldoende om het organisme te versterken.
Overbelasting en overbelasting dienen in principe te worden vermeden. Alle bewegingen moeten worden aangepast aan de huidige fysieke mogelijkheden. Aangezien deze veranderen in de loop van de ziekte, moeten ze regelmatig worden aangepast aan de omstandigheden. De patiënt mag zichzelf nooit overbelasten of blootstellen aan een groot valrisico. Eenzijdige belasting, een slechte lichaamshouding of een starre lichaamshouding moeten bij alle bewegingssequenties worden vermeden. Het skeletstelsel heeft balansposities nodig en daarnaast voldoende warmtetoevoer.
Als de patiënt een slecht gezichtsvermogen heeft of blind is, moeten het dagelijkse leven en de ruimtelijke omstandigheden worden aangepast aan zijn behoeften. Het risico op ongevallen moet worden geminimaliseerd en er is dagelijkse hulp nodig voor het uitvoeren van verschillende taken.
Gesprekken met psychologen, familieleden en vrienden kunnen belangrijk zijn voor mentale versterking. Bovendien vinden veel patiënten deelname aan zelfhulpgroepen prettig en nuttig. De uitwisseling met andere zieke mensen biedt wederzijdse ondersteuning. Ervaringen kunnen worden besproken en tips worden gegeven om in het dagelijks leven met de ziekte om te gaan.
.jpg)
.jpg)










.jpg)