De Baller-Gerold-syndroom behoort tot de groep van misvormingssyndromen waarbij het gezicht voornamelijk is aangetast. Het syndroom is te wijten aan mutaties en wordt overgedragen op een autosomaal dominante wijze van overerving. De therapie is beperkt tot symptomatische behandeling, die grotendeels bestaat uit chirurgische correctie van de misvormingen.
Wat is het Baller-Gerold-syndroom?

© adimas - stock.adobe.com
In de ziektegroep van congenitale misvormingssyndromen vormen misvormingssyndromen met overwegend gelaatstrekken een aparte subgroep. Baller-Gerold-syndroom valt in deze subgroep. De ziekte dankt zijn naam aan de eerste beschrijvingen van F. Baller en M. Gerold, die de ziekte voor het eerst in het midden van de 20e eeuw documenteerden.
De prevalentie die voor het syndroom wordt gegeven, is een verhouding van één op 1.000.000. De belangrijkste symptomen van het symptoomcomplex zijn de vroege sluiting van de craniale hechtingen in de zin van craniosynostose en het niet plaatsen van de radiale botten. Volgens de huidige kennis is het Baller-Gerold-syndroom een erfelijke aandoening die kan worden herleid tot een genetische mutatie.
oorzaken
Hoewel het Baller-Gerold-syndroom uiterst zeldzaam is, is de oorzaak al bekend. Een genetische mutatie in het RECQL4-gen op chromosoom 8q24.3 is verantwoordelijk voor het syndroom. Het gen in menselijk DNA codeert voor een enzym genaamd RecQ Helicase. Dit enzym ontwart de DNA-strengen en bereidt ze voor op reduplicatie. Als het coderende gen gemuteerd is, is het enzym defect en kan het zijn taak niet meer naar tevredenheid uitvoeren.
Het resultaat zijn de individuele symptomen van het Baller-Gerold-syndroom. In verband met de oorzakelijke mutatie werd familiaire accumulatie waargenomen, wat spreekt voor de erfelijke aard van de ziekte. Blijkbaar wordt de mutatie overgedragen op de autosomaal dominante wijze van overerving. Het is nog niet helemaal duidelijk of naast genetische factoren ook externe factoren zoals blootstelling aan toxines een rol spelen bij het ontstaan van de ziekte.
Symptomen, kwalen en tekenen
Klinisch gezien wordt het symptoomcomplex van het Baller-Gerold-syndroom gekenmerkt door voortijdige hechtingssynostose, die zelfs vóór de geboorte plaatsvindt. De patiënten lijden ook aan een radiaal defect dat geassocieerd is met aplasie van de radius en soms met een misvorming van de duim.
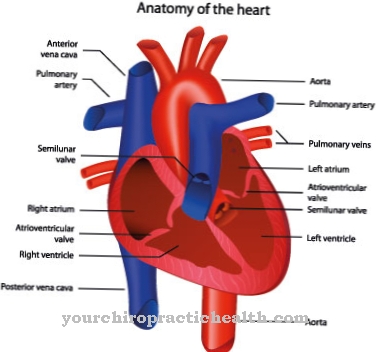
De pols en metacarpale botten aan de duimzijde kunnen ook dysplastisch of hypoplastisch lijken. Hetzelfde geldt voor de daar aanwezige vinger-extensoren. Bovendien lijden de patiënten vaak aan een kleine gestalte, wat gepaard kan gaan met skeletmisvormingen van de wervelkolom of misvormingen van de schouder- en bekkengordels. Sommige mensen hebben ook hartafwijkingen, anale atresie of ontwrichte nieren.
Vaak komen ook gezichtsdysmorfismen voor, die zich kunnen uiten in de vorm van hypertelorisme, een epicanthus of een prominente wortel van de neus. Misvormde oorschelpen zijn ook denkbare dysmorfismen. Vaak zijn de getroffenen mentaal gehandicapt vanwege de vroege naadsluiting. Als zuigeling lijden ze soms aan poikilodermie.Bij kinderen manifesteert de ziekte zich vaak in de vorm van hypoplasie van de patella. De mutatie maakt patiënten ook vatbaarder voor het ontwikkelen van osteosarcomen.
Diagnose en verloop
Bij het diagnosticeren van het Baller-Gerold-syndroom moet de arts het klinische beeld na de eerste visuele diagnose onderscheiden van klinisch vergelijkbare syndromen zoals het Rapadilino-syndroom of het Rothmund-Thomson-syndroom. Deze syndromen zijn gebaseerd op dezelfde genetische mutatie. Een andere oorzaak is het klinisch relatief vergelijkbare Saethre-Chotzen-syndroom, dat ook moet worden gedifferentieerd.
Bovendien moeten de radiale defecten van het syndroom worden onderscheiden van het Roberts-syndroom. Als poikilodermie aanwezig is, wordt dit symptoom pathognomonisch genoemd. Theoretisch is er de mogelijkheid van een moleculair genetische analyse voor diagnose en bevestiging van de diagnose. De prognose hangt af van de manifestaties in het individuele geval. Op de lange termijn verslechteren bijvoorbeeld osteosarcomen de prognose aanzienlijk.
Complicaties
Het Baller-Gerold-syndroom kan leiden tot verschillende complicaties die kunnen leiden tot verkeerde posities in het lichaam. Het Baller-Gerold-syndroom leidt in de regel tot misvormingen van het skelet en de wervelkolom. Deze misvormingen gaan gepaard met een kleine gestalte en hebben een zeer negatief effect op de kwaliteit van leven.
Vooral kinderen kunnen last hebben van een klein postuur als ze vanwege het symptoom worden geplaagd of gepest. Het is niet ongebruikelijk dat het Baller-Gerold-syndroom gepaard gaat met mentale retardatie, wat de concentratie en het leervermogen van de patiënt aanzienlijk vermindert. De betrokkene is vaak afhankelijk van de hulp en zorg van andere mensen.
De symptomen verminderen de levensverwachting. Ook neemt de kans op tumorvorming toe, wat kan leiden tot levensbedreigende complicaties die in het ergste geval tot de dood kunnen leiden. De causale behandeling van het Baller-Gerold-syndroom vindt meestal direct na de geboorte plaats door middel van chirurgische ingrepen.
Verder moet de patiënt voorzichtig zijn met het gebruik van veel zonbescherming zodat huidkanker niet optreedt. Het Baller-Gerold-syndroom kan leiden tot loopstoornissen en verdere breuken die het leven moeilijk kunnen maken. De symptomen kunnen echter worden beperkt met behulp van loophulpmiddelen.
Wanneer moet je naar de dokter gaan?
Als het Baller-Gerold-syndroom en soortgelijke syndromen worden vermoed, moet een doktersbezoek worden gebracht. Na de eerste visuele diagnose kan de arts de ziekte onderscheiden van vergelijkbare ziekten en de symptomen gaan behandelen. Typische waarschuwingssignalen die medische opheldering vereisen, zijn misvormingen van het handwortel- en middenhandsbeentje aan de duimzijde, vooral van de duim en strekspieren. Bovendien zijn er meestal misvormingen van de wervelkolom en schouder- of bekkengordel, evenals een korte gestalte.
Hartafwijkingen en misvormde wortels van de neus, oorschelpen en jukbeenderen kunnen ook voorkomen. Als een van deze symptomen wordt gevonden, moet een arts worden geraadpleegd. Meestal wordt de ziekte echter in de kindertijd vastgesteld. Getroffen kinderen zijn meestal verstandelijk gehandicapt en worden getest op het Baller-Gerold-syndroom als onderdeel van het eerste onderzoek na de geboorte.
Als het syndroom zwak is, wordt de diagnose meestal in de eerste levensjaren gesteld. Als het bovengenoemde ziektebeeld optreedt, moet het betreffende kind zeker naar de kinderarts worden gebracht. Dit is vooral het geval als in het verleden lichte afwijkingen of mentale problemen zijn opgemerkt.
Artsen & therapeuten bij u in de buurt
Behandeling en therapie
Causale therapie is niet beschikbaar voor patiënten met het Baller-Gerold-syndroom. Gentherapeutische benaderingen zijn momenteel het belangrijkste onderwerp van onderzoek in de geneeskunde. Ze zijn echter nog niet in de klinische fase gekomen. Om deze reden zijn alle ziekten die op genetische mutaties zijn gebaseerd tot dusverre ongeneeslijk. Totdat gentherapiebenaderingen zijn goedgekeurd, wordt een puur symptomatische behandeling voor dergelijke ziekten gebruikt.
Bij misvormingssyndromen zoals het Baller-Gerold-syndroom komt symptomatische behandeling meestal overeen met het chirurgisch oplossen van de meervoudige misvormingen. De craniosynostose van de patiënt wordt meestal binnen de eerste zes maanden chirurgisch gecorrigeerd, zodat de daaropvolgende symptomen zo laag mogelijk blijven. De duim kan eventueel chirurgisch reconstructief worden behandeld.
Deze behandeling is meestal gebaseerd op een chirurgische transpositie van de wijsvinger. Aanwezige hartafwijkingen moeten zo snel mogelijk worden gecorrigeerd. In individuele gevallen kan chirurgische correctie van verplaatste nieren ook nodig zijn. Mentale retardatie kan ondersteunend worden bestreden door vroege interventiemethoden. In de regel ontwikkelen kinderen die op deze manier worden ondersteund, ondanks het syndroom een normale intelligentie.
Vanwege hun aanleg voor kanker moeten patiënten met het Baller-Gerold-syndroom ook regelmatig en zo nauwkeurig mogelijk deelnemen aan preventieve onderzoeken. Mensen wordt vaak geadviseerd om blootstelling aan de zon te vermijden. Dit vermindert de gevoeligheid voor kankers zoals huidkanker.
Wanneer kanker uitbreekt, wordt de ziekte meestal vroeg genoeg herkend door middel van hechte preventieve onderzoeken. In een groot aantal gevallen leiden de getroffenen op volwassen leeftijd een relatief normaal leven en genieten ze nauwelijks een verminderde kwaliteit van leven.
preventie
Preventieve maatregelen voor genetische en mutatiegerelateerde ziekten zoals het Baller-Gerold-syndroom zijn slechts met mate beschikbaar. Nadat bewijs van pathogene RECQL4-mutaties is vastgesteld, kunnen getroffen ouders prenatale diagnostiek gebruiken in het geval van verdere zwangerschappen en, onder bepaalde omstandigheden, een beslissing nemen tegen het kind. Erfelijkheidsadvies bij gezinsplanning kan ook grotendeels worden omschreven als een preventieve maatregel.
U kunt dat zelf doen
De patiënten met het Baller-Gerold-syndroom lijden aan misvormingen van sommige inwendige organen die chirurgische correctie vereisen. Zelfhulpmaatregelen zijn bij deze klachten niet mogelijk, maar de betrokkene ondersteunt het succes van medische therapieën door middel van hun gedrag. Tijdens zijn verblijf in de kliniek volgt hij de instructies van de doktoren op en ondersteunt hij de regeneratie van het organisme met voldoende rust.
De hand en vingers worden vaak aangetast door misvormingen die dagelijkse manipulaties bemoeilijken. De patiënt bevordert zijn motorische vaardigheden op de juiste gebieden door fysiotherapie te volgen en de thuis geleerde trainingseenheden uit te voeren. Een soortgelijke therapie kan ook worden toegepast bij eventuele loopstoornissen, waarbij de patiënt vaak ook loophulpmiddelen gebruikt.
Sportactiviteiten zijn mogelijk afhankelijk van de fysieke conditie van de betrokkene en hebben doorgaans een gunstig effect op het welzijn. Voorwaarde is echter dat alle sportactiviteiten zijn goedgekeurd door een arts.
Als de ziekte gepaard gaat met verminderde cognitieve prestaties, krijgen patiënten nog steeds waardevol onderwijs op scholen voor speciaal onderwijs. De sociale contacten die het gevolg zijn van het naar school gaan, verhogen ook de kwaliteit van leven van de getroffenen. Als de ouders van de zieke kinderen door de extra stress een depressie of burn-out krijgen, wenden zij zich zo snel mogelijk tot een psychologische therapeut.

.jpg)
.jpg)
.jpg)


.jpg)