Bij de Apert-syndroom het is een zeldzame erfelijke ziekte. Misvormingen komen voor in het hele organisme met de meest ernstige, chronische functionele verliezen en het verloop van de ziekte door misvormingen. Beide geslachten worden in gelijke mate beïnvloed.
Wat is het apert-syndroom?

© Sashkin - stock.adobe.com
Het Apert-syndroom, ook wel bekend als Acrocefalosyndactyliesyndroom waarvan bekend is dat ze de oorzaak zijn van ernstige tot zeer ernstige misvormingen die het hele lichaam kunnen treffen. Deze meervoudige misvormingen werden voor het eerst beschreven door de Franse kinderarts Eugène Apert, naar wie de ongeneeslijke ziekte ook werd genoemd. Eugène Apert werkte als kinderarts in de Franse hoofdstad Parijs en leefde van 1868 tot 1940.
Het Apert-syndroom is slechts een van de mogelijke andere fenotypische manifestaties van het acrocefalosyndactyliesyndroom. In het ziekteregister van de ICD-10 heeft het Apert-syndroom de diagnosecode Q87.0. De 4 andere ziekten, die ook deel uitmaken van de acrocefalosyndactyliesyndromen, zijn onder meer het Crouzon-syndroom, het Carpenter-syndroom, het Pfeiffer-syndroom en het Saethre-Chotzen-syndroom. Het Pfeiffer-syndroom moet niet worden verward met infectieuze mononucleosis, ook bekend als de ziekte van Pfeiffer.
Onder de genoemde ziekten van de acrocefalosyndactyliesyndromen is het Apert-syndroom echter degene met het meest ernstige en fulminante verloop. Nadat de vermoedelijke diagnose is gesteld, staan de artsen voor de uitdaging om het klinische beeld toe te wijzen aan de juiste diagnose uit de groep van acrocefalosyndactyliesyndromen.
oorzaken
Alle acrocefalosyndactyliesyndromen, inclusief het Apert-syndroom, zijn gebaseerd op een speciale genmutatie. Moleculair-biologische tests hebben inmiddels aangetoond dat het fenotype van het Apert-syndroom, d.w.z. het uiterlijk zichtbare uiterlijk van de ziekte, terug te voeren is op een mutatie van het 10e chromosoom. Deze verbanden waren toen echter nog niet bekend bij de ontdekker van de ziekte. Op chromosoom 10 staat de blauwdruk voor het zogenaamde fibroblastgroeifactorreceptorgen, FGFR.
De exacte genlocus is 10Q26, het Apert-synroom wordt geërfd via een zogenaamde autosomaal dominante overerving. Als het overeenkomstige gen op chromosoom 10 is gemuteerd, dan is er altijd sprake van een fenotypische uitbraak van de ziekte; in deze context spreekt men van volledige penetrantie. De variantie bij het Apert-syndroom is erg hoog; dit is van klinische relevantie in zoverre de tekenen en symptomen van de ziekte bij de getroffenen heel verschillend kunnen zijn. De meest voorkomende oorzaken van het Apert-syndroom zijn spontane, d.w.z. onvoorspelbare mutaties op het 10e chromosoom.
Symptomen, kwalen en tekenen
Moleculair biologisch onderzoek heeft zonder enige twijfel aangetoond dat de leeftijd van de vader van het kind een belangrijke rol speelt in de neiging tot spontane mutatie. Het Apert-syndroom komt daarom vooral voor bij kinderen van oudere vaders. Deze relatie is geverifieerd voor het Apert-syndroom, maar niet voor de andere acrocefalosyndactyliesyndromen. Statistisch gezien wordt één op de 130.000 pasgeborenen getroffen door het Apert-syndroom. De symptomen, klachten en tekenen die op de aanwezigheid van een Apert-syndroom kunnen duiden, zijn zeer divers en variabel.
Bij een volledig ontwikkeld ziektebeeld wordt de juiste diagnose betrouwbaar gesteld, met subformulieren kan er echter ook vertraging optreden bij de juiste diagnose. De hoofdsymptomen van het Apert-syndroom zijn misvormingen van de benige schedel, het gezicht, ledematen inclusief handen en voeten, evenals scoliose van de wervelkolom, ademhalingsproblemen, ametropie en gehoorverlies.
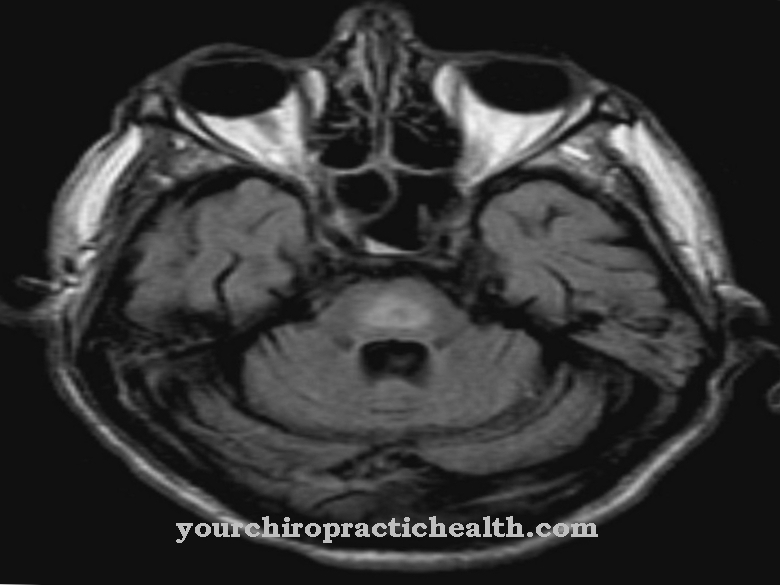
In meer dan 80 procent van de gevallen kan de arts ook vaststellen wat bekend staat als hydrocephalus en ernstige mentale retardatie. De botten van de schedel zijn zo versmolten dat kneuzingen van de hersenen en dus verhoogde intracraniale druk optreden.
Diagnose en verloop
Typische misvormingen en kenmerken van een Apert-syndroom zijn vanaf ongeveer de vierde maand van de zwangerschap te herkennen aan fijne echografie. Direct na de geboorte kan op basis van de typische indicatieve symptomen een eerste vermoedelijke diagnose worden gesteld. De diagnose moet onmiddellijk worden bevestigd en onderbouwd door verdere moleculair genetisch bloedonderzoek. Als de diagnose dan onomstotelijk vaststaat, moet de chirurgische therapie onmiddellijk worden gestart.
Complicaties
Bij het Apert-syndroom zijn er zeer ernstige complicaties die het leven van de patiënt ernstig beïnvloeden. Het Apert-syndroom veroorzaakt ernstige misvormingen op het lichaam die tot gezondheidsproblemen kunnen leiden. Zowel vrouwen als mannen worden getroffen door het syndroom.
In de regel verschijnt het syndroom als een misvorming in het schedelbot. De schedel ziet er daardoor anders uit dan bij gezonde mensen. Dit leidt vaak tot pesten en plagen, vooral bij kinderen, en kan bij patiënten psychische problemen veroorzaken. De ledematen kunnen ook misvormingen bevatten, en er zijn ook ademhalingsproblemen en gehoorverlies.
De meeste patiënten hebben een visueel hulpmiddel nodig. Naast fysieke en fysieke problemen ervaren de meeste patiënten ook mentale en psychische problemen. Het denken en handelen wordt ernstig beperkt, zodat de patiënt in de meeste gevallen het leven van alledag niet alleen aankan en ook afhankelijk is van de hulp van de familie.
Behandeling is slechts in beperkte mate mogelijk. De lichamelijke symptomen kunnen worden behandeld met chirurgische ingrepen, zodat misvormingen en verkeerde uitlijningen worden verwijderd. De mentale retardatie is echter niet te behandelen, zodat de patiënten ook een kortere levensverwachting hebben.
Wanneer moet je naar de dokter gaan?
Bij het Apert-syndroom is een vroege diagnose en behandeling van de ziekte van groot belang. Om deze reden moet een arts worden geraadpleegd als de symptomen van dit syndroom optreden. De getroffenen lijden in de regel aan verschillende misvormingen van het lichaam, die echter niet direct na de geboorte hoeven op te treden. Daarom moet een medisch onderzoek worden uitgevoerd als de patiënt moeite heeft met ademhalen of zien. Een onderzoek en behandeling door een arts is ook zinvol bij gehoorverlies dat zonder specifieke reden optreedt.
Het is niet ongebruikelijk dat patiënten lijden aan een verhoogde vertraging als gevolg van het Apert-syndroom. Dit kan zich uiten door moeilijkheden op school en door mentale en motorische beperkingen. Als deze symptomen ook optreden, is de kans groot dat de patiënt het Apert-syndroom heeft.
Met vroege behandeling kunnen de symptomen worden beperkt, zodat het dagelijks leven draaglijk is voor de betrokkene. Veel symptomen kunnen worden opgelost of verlicht door verschillende chirurgische ingrepen. Een bezoek aan de dokter kan ook nuttig zijn voor de ouders of familieleden van het kind als zij last hebben van psychische klachten of depressies.
Artsen & therapeuten bij u in de buurt
Behandeling en therapie
De focus van de therapie ligt op het verwijderen van de schedelmisvormingen die verantwoordelijk zijn voor de toename van de intracraniale druk bij hydrocephalus. De hiervoor benodigde therapeutische ingreep kan alleen operatief worden uitgevoerd. Om dit te doen, moeten de craniale hechtingen, de hechtingen, onder algemene anesthesie worden gestraald. Deze speciale vorm van chirurgie kan alleen worden uitgevoerd in gekwalificeerde centra.
De verkeerde uitlijning van tenen en vingers wordt meestal zoveel mogelijk synchroon uitgevoerd. In een verdere, aparte procedure moeten structuren in het middenoor geopereerd worden omdat de typische gehoorbeentjes, hamer, aambeeld en stijgbeugel niet of niet volledig ontwikkeld zijn bij het Apert syndroom.
In sommige gevallen kan deze operatie kinderen helpen om ten minste een deel van hun gehoor terug te krijgen. Als de misvormingen in het middenoor te uitgesproken zijn, kan het gehoor helaas niet worden hersteld. De getroffen kinderen worden blootgesteld aan zeer extreme psychologische stress, omdat de afwijzing door de omgeving erg hoog is. En de getroffen kinderen ondergaan in hun eerste levensjaren al een groot aantal operaties. De psychologische beperkingen die daardoor worden verergerd, moeten altijd ook worden behandeld.
Outlook & prognose
De prognose voor het Apert-syndroom moet als ongunstig worden geclassificeerd. De erfelijke ziekte komt vaker voor bij kinderen met een oudere vader en wordt tot op de dag van vandaag nog als ongeneeslijk beschouwd. Om juridische redenen is het artsen en wetenschappers verboden zich te mengen in de menselijke genetica. Daarom is er met de momenteel beschikbare opties geen therapie of medicijn dat zou kunnen leiden tot herstel van de ziekte.
Het behandelplan is gericht op het verlichten van het bestaande ongemak en richt zich op het verbeteren van de kwaliteit van leven. In veel gevallen kan de behandeling echter niet worden uitgevoerd omdat het geen verder succes zou hebben. Hoewel de ziekte ongeneeslijk is, is ze niet progressief. De bestaande beperkingen verergeren in de loop van het leven niet.
Visiehulpmiddelen worden tijdens de behandeling vaak gebruikt om het gezichtsvermogen te verbeteren. Bovendien wordt, indien mogelijk, een operatie uitgevoerd om de misvormingen van de schedel te veranderen. Dit heeft niet alleen een cosmetisch karakter, maar zorgt er ook voor dat bloedvaten en weefsel tijdens het groeiproces niet verpletterd of beschadigd raken. Dit zijn vaak preventieve maatregelen zodat er geen acuut orgaanfalen of bloeding kan optreden. Dit zou leiden tot vroegtijdig overlijden van de patiënt.
preventie
Directe preventie van spontane mutaties is nog niet mogelijk. Het acrocefalosyndactyliesyndroom kan echter vroeg in een zogenaamde prenatale diagnose worden opgespoord. Vanaf de negende week van de zwangerschap is ook een vruchtwaterpunctieonderzoek mogelijk, waarmee de diagnose al kan worden gesteld. Als acrocefalosyndactyliesyndroom waarschijnlijk is, zullen artsen u gewoonlijk adviseren om de zwangerschap zo snel mogelijk te onderbreken.
Nazorg
Apert-syndroom wordt altijd geassocieerd met langdurige therapie. De zieken hebben maanden of jaren medische en therapeutische ondersteuning nodig, aangezien de verschillende klachten en symptomen in chronische vorm kunnen terugkeren of aanhouden. Nazorg richt zich dan ook op regelmatige vervolgcontroles.
Patiënten dienen in het begin wekelijks een arts te raadplegen. Als de ziekte positief verloopt, kunnen de controles geleidelijk worden afgebouwd, waarbij de routineonderzoeken permanent moeten worden uitgevoerd. Als zich complicaties voordoen tijdens de follow-up, moet de behandeling worden hervat.
Patiënten moeten ook de medicatie zoeken die ze nodig hebben om effectief bijholteontstekingen, ademhalingsmoeilijkheden en andere, soms chronische, symptomen te verlichten. Nazorg omvat ook het veranderen van bepaalde leefgewoonten, waardoor de symptomen kunnen verergeren. Dus het dieet moet worden veranderd.
Een vetarm en vitaminerijk dieet kan ontstekingen verminderen en het immuunsysteem ondersteunen. Vanwege de verschillende trajecten die de genetische aandoening kan doorlopen, moeten de exacte stappen steeds worden uitgewerkt met het oog op het individuele symptoomprofiel van de patiënt. De verantwoordelijke arts is de juiste contactpersoon voor nazorg na het Apert-syndroom.
U kunt dat zelf doen
Apert-syndroom is een ernstige ziekte die gepaard gaat met een aantal klachten en aanzienlijke fysieke en psychische stress voor de getroffenen. De getroffenen moeten daarom profiteren van therapeutische maatregelen die conventionele medische behandelingen ondersteunen.
Uitwisseling met andere getroffen personen is mogelijk als onderdeel van een zelfhulpgroep. Hierdoor kun je niet alleen tips uitwisselen over hoe om te gaan met de ziekte, maar ook nieuwe contacten leggen met andere zieke mensen en specialisten. Op de lange termijn moet de ziekte echter worden geaccepteerd - door de getroffenen en hun familieleden tegelijk. Het ouderinitiatief Apert Syndrome and Related Malformations e.V. biedt de getroffenen verdere contactmogelijkheden en tips om op een gezonde manier met de ziekte om te gaan.
Afgezien van deze therapeutische maatregelen, kunnen de symptomen ook worden verlicht door een gezonde levensstijl. Regelmatige lichaamsbeweging en gezonde voeding helpen op de lange termijn om de symptomen te beheersen en de kwaliteit van leven te verbeteren. Bovendien moeten de nodige maatregelen worden genomen om het toekomstige leven toegankelijk te maken voor mensen met een handicap. Genoemde maatregelen kunnen het beste worden uitgevoerd in overleg met de verantwoordelijke arts en naasten.
.jpg)

.jpg)

.jpg)



.jpg)