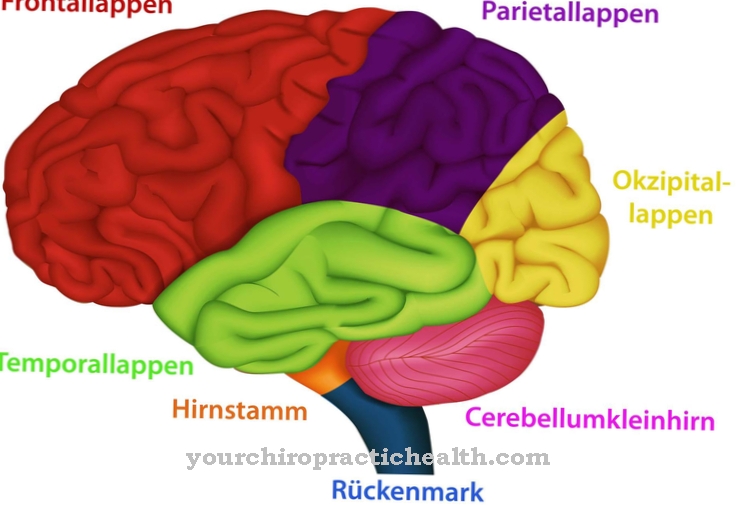
De Bardet-Biedl-syndroom, ook Syndroom van Laurence-Moon-Biedl-Bardet (LMBBS), is een ziekte op het gebied van ciliopathieën die uitsluitend optreedt als gevolg van erfelijkheid. Het syndroom manifesteert zich in de vorm van meerdere misvormingen die worden veroorzaakt door veranderingen (mutaties) op verschillende genlocaties of chromosomen.
Wat is het Bardet-Biedl-syndroom?

© Creativa afbeeldingen - stock.adobe.com
Het klinische beeld gedefinieerd door de artsen Moon en Laurence en later door Bardet en Biedl is een ziekte waarbij retinale dystrofie optreedt als medisch significant kenmerk in combinatie met andere symptomen. Door deze ingewikkelde medische uitgangssituatie is de uiteindelijke vaststelling van de ziekte van BBS moeilijk. Dit ziektebeeld werd voor het eerst medisch vastgelegd in 1866.
Vier onderzochte personen hadden retinitis pigmentosa (retinale dystrofie, RP) in combinatie met een dwarslaesie (spastische verlamming), hypogenitalisme (onderontwikkelde geslachtsorganen) en een verstandelijke handicap. In 1920 beschreef de Franse arts Bardet een ziekte die bestond uit RP (retinale dystrofie), hypogenitalisme, polydactylie en obesitas.
De Praagse patholoog Biedl ontdekte ook zwakte (mentale verwarring). In 1925 vatten de onderzoekers Weiss en Solis-Cohen de bekende gevallen samen en beschreven het klinische beeld als Syndroom van Laurence-Moon-Biedl-Bardet.
oorzaken
In de jaren die volgden, wees de medische literatuur er steeds meer op dat de gevallen geregistreerd door Laurence en Moon een zeldzame bijzondere vorm zijn die alleen in individuele gevallen samen met BBS voorkomt. Meer recente medische onderzoeksresultaten wijzen het Bardet-Biedl-syndroom toe aan het gebied van ciliopathieën (ciliaire aandoeningen).
Deze ziekten vertonen een veel voorkomende storing van de zogenaamde cilia (kleine processen, antennes), die voorkomen op de meeste cellen van het menselijk organisme. De ciliopathieën worden gekenmerkt door vloeiende overgangen en overlappingen tussen verschillende ciliaire aandoeningen.
Symptomen, kwalen en tekenen
Het belangrijkste kenmerk van erfelijke retinale dystrofie is een algemene term die het begin van functieverlies en de daaropvolgende degeneratie (vernietiging) van de fotoreceptoren beschrijft. Ze leiden tot een progressief (progressief) verlies van visuele functie. De snel voortschrijdende visuele stoornissen treden meestal heel vroeg op bij kinderen tussen de vier en tien jaar oud. Ze laten zich op verschillende manieren voelen, afhankelijk van de getroffen fotoreceptoren.
Als een "staafvormige kegelvorm" met het karakteristieke verloop van retinitis pigmentosa (RP), vindt de ziekte zijn oorsprong in de retinale periferie (buitenste retina) en ontwikkelt zich tot maculaire degeneratie (vernietiging van scherp zicht) via een progressief verlies van gezichtsveld.
Bij obesitas (obesitas) vertoont het lichaam een pathologische ophoping van vetweefsel. In het geval van BBS treden abnormaal verhoogde vetophopingen op de benen, buik, billen, armen, borst en heupen voornamelijk op als rompobesitas, waarbij de romp, benen en dijen bijzonder zwaar worden aangetast. Polydactylie is een merkbaar symptoom en een significant kenmerk van het Bardet-Biedl-syndroom. De bevinding is niet eenvoudig omdat de rudimentaire polydactylie na de geboorte operatief wordt gecorrigeerd.
Röntgenfoto's kunnen meer informatie geven. Polydactylie kan met verschillende tekens verschijnen, bijvoorbeeld als een rudimentair teen- of vingeraanhangsel. Een teen of vinger kan aanvullend of slechts gedeeltelijk worden gevormd. De eenzijdige hexadactylie aan de voet en / of hand heeft een extra schakel, de bilaterale hexadactylie komt voor op beide voeten en / of handen.
Tenen of vingers die samengegroeid zijn (syndactylie) en verkorting van een of meer tenen of vingers (brachydactylie) zijn ook tekenen van BBS. Slechts een paar patiënten hebben alle vier de aangedane ledematen. De mentale ontwikkelingsachterstand is anders. Slechts een klein aantal getroffenen vertoont ernstige mentale retardatie. Een normaal getrainde intelligentie is mogelijk.
Kinderen leren laat spreken en lopen, en vertonen soms gedragsproblemen zoals angststoornissen. Dwangmatig of autistisch gedrag, een lage drempel voor frustratie en onstabiele emotionaliteit zijn andere mogelijke bijwerkingen. Het bekende heeft de voorkeur, maar wijzigingen worden afgewezen. Afwijkingen in de interne en externe geslachtsorganen komen vaak voor.
Andere veranderingen zijn hypospadie (de urethrale opening is boven of onder, in plaats van aan de voorkant van de penis), abdomen of lies testikels, urethrale vernauwingen, voorhuid vernauwing en posterieure urethrale kleppen. Bij vrouwelijke patiënten zijn vaginale atresie (vagina is niet doorheen open), ontbrekende urethrale openingen en verminderde binnenste schaamlippen bekend.
Het is niet ongebruikelijk dat de getroffen vrouwen een onregelmatige menstruatiecyclus hebben. Nierveranderingen zijn vaak voorkomende bijwerkingen. De bevinding hangt af van het onderzoek van de lagere urinewegen en de nieren met behulp van echografie (echografie).
Diagnose en ziekteverloop
Het Bardet-Biedl-syndroom (BBS) heeft zes hoofdsymptomen, maar ze komen niet altijd samen voor. Artsen gaan uit van een overeenkomstige bevinding als er ten minste vier van de belangrijkste symptomen aanwezig zijn. Als alternatief is de kans groot dat de ziekte aanwezig is als de patiënt drie hoofdsymptomen en twee secundaire symptomen heeft.
De zes belangrijkste symptomen zijn retinale dystrofie, obesitas (abnormale ophoping van vetweefsel, overgewicht), polydactylie (overtollige tenen en / of vingers), mentale retardatie (mentale ontwikkelingsachterstand), hypogenitalisme (onderontwikkelde geslachtsorganen) en nierziekte. Laagfrequente secundaire symptomen zijn onder meer spraakvertragingen, spraakstoornissen, hartafwijkingen, ataxie (verminderde bewegingscoördinatie), astma, diabetes mellitus (diabetes), de ziekte van Crohn (ontsteking van de dikke en / of dunne darm), dysplasie van de ribben en wervels en kyfoscoliose (vertebrale anemie) Aan.
Complicaties
Met het Laurence-Moon-Biedl-Bardet-syndroom lijden de getroffenen meestal aan een verlies van visuele functie. Het verlies treedt niet plotseling op, maar geleidelijk. In het ergste geval worden de getroffenen volledig blind, wat meestal niet meer te behandelen is.
Vooral bij jongeren en kinderen kan blindheid leiden tot ernstige psychische klachten of zelfs depressie. De patiënten zijn duidelijk beperkt in hun dagelijks leven en hebben een sterk verminderd gezichtsveld. In veel gevallen leidt het Laurence-Moon-Biedl-Bardet-syndroom ook tot gedragsproblemen, waardoor met name kinderen last kunnen hebben van pesten of plagen.
De ontwikkeling van kinderen wordt ook aanzienlijk vertraagd en beperkt door het syndroom. Angststoornissen kunnen ook voorkomen. Het is niet ongebruikelijk dat het Laurence-Moon-Biedl-Bardet-syndroom leidt tot psychische klachten en depressies bij familieleden of ouders. Een causale behandeling van het Laurence-Moon-Biedl-Bardet-syndroom is helaas niet mogelijk.
Sommige klachten kunnen beperkt zijn. Een volledig positief verloop van de ziekte zet echter niet in. Het syndroom vermindert de levensverwachting van de patiënt niet. In sommige gevallen hebben getroffenen soms hulp van andere mensen nodig in hun dagelijks leven.
Wanneer moet je naar de dokter gaan?
Aangezien het Laurence-Moon-Biedl-Bardet-syndroom een erfelijke ziekte is, kan de diagnose in de baarmoeder worden gesteld. Uiterlijk na de bevalling moet een arts worden geraadpleegd als typische symptomen zoals gezichtsstoornissen of zwaarlijvigheid worden opgemerkt. Misvormingen van de tenen en vingers zijn ook een duidelijke indicator van een ziekte.Ouders die symptomen bij hun kind opmerken, moeten de kinderarts onmiddellijk informeren.
Een uitgebreid onderzoek geeft informatie over de ziekte. Daarna wordt meestal direct gestart met de therapie, die bestaat uit verschillende behandelingen door orthopedisten, neurologen, oogartsen, internisten en therapeuten en fysiotherapeuten. Verdere bezoeken aan de arts zijn nodig als de behandeling niet het gewenste effect heeft. Medisch advies is ook nodig in noodsituaties, bijvoorbeeld als het kind valt als gevolg van een misvorming of plotseling een aanval krijgt. Als de zieke tekenen van emotioneel ongemak vertoont, moeten de ouders een geschikte therapeut raadplegen. Oudere kinderen kunnen samen met hun ouders contact opnemen met de schoolpsycholoog en passende maatregelen bespreken.
Therapie en behandeling
Deze ziekte ontstaat op basis van autosomaal recessieve overerving, wat betekent dat beide kopieën (allelen) van een BBS-gen een verandering (mutatie) vertonen. De ouders van de patiënt zijn 'gemengdbloedig' en dragen elk een gemodificeerd en een ongewijzigd allel van het overeenkomstige gen. Ze hebben de ziekte niet. Kinderen worden pas ziek als hun vader en moeder het gemuteerde allel doorgeven. Bij extra kinderen is de kans op herhaling 25 procent.
Een causale therapieoptie is nog niet bekend, aangezien bepaalde symptomen van de ziekte nog niet definitief kunnen worden toegewezen aan de verschillende genetische veranderingen. De symptomen en hun manifestaties verschijnen anders, zelfs bij zieke broers en zussen. Omdat het kenmerkende volledige beeld van BBS slechts in zeldzame gevallen aanwezig is, vooral bij kleine kinderen, is een overeenkomstige diagnose moeilijk.
Vanwege de veelvuldig aanwezige oligosymptomatische symptomen, waarbij zeer weinig atypische en slechts licht uitgesproken symptomen optreden, moet bij de differentiële diagnose rekening worden gehouden met andere mogelijke ziektebeelden. Veranderingen in hetzelfde gen kunnen leiden tot verschillende ziektebeelden, bijvoorbeeld het Joubert-, Bardet-Biedl- of Meckel-Gruber-syndroom.
Outlook & prognose
De prognose voor de aanwezigheid van het Laurence-Moon-Biedl-Bardet-syndroom is over het algemeen slecht omdat de meervoudige misvormingen aangeboren en ongeneeslijk zijn. Als vier van de zes hoofdsymptomen optreden, wordt de diagnose van het Laurence-Moon-Biedl-Bardet-syndroom bevestigd. Talrijke secundaire symptomen dragen bij aan de belangrijkste symptomen. Dit omvat een sluipende blindheid.
Vanwege de complexiteit van de symptomen is er geen uitzicht op genezing. Er is slechts een middelmatige kans op merkbare symptoomverlichting. Het aantal mogelijke misvormingen en aandoeningen bij het Laurence-Moon-Biedl-Bardet-syndroom is zo groot dat de erfelijke ziekte moeilijk te behandelen is. Het beloop van deze erfelijke ziekte is in ieder geval niet te beïnvloeden. De huidige symptomen kunnen echter gedeeltelijk worden verlicht.
De slechte algehele prognose vermindert echter de levensverwachting van de getroffen mensen niet. Op gevorderde leeftijd en nadat ze blind zijn geworden, kunnen de getroffenen permanent afhankelijk zijn van hulp of zorg. Door interdisciplinaire medische inspanningen kunnen veel patiënten met het Laurence-Moon-Biedl-Bardet-syndroom een iets milder verloop van de ziekte ervaren.
De toenemende visuele problemen vormen een moeilijk te behandelen en problematisch deel van de ziekte. Toenemende visuele beperkingen treden al op bij de getroffen kleine kinderen. Ze verslechteren na verloop van tijd. De zichtproblemen hoeven niet bij alle getroffenen tot blindheid te leiden. De psychologische gevolgen van het Laurence-Moon-Biedl-Bardet-syndroom kunnen meestal goed worden behandeld.
preventie
Preventie in de zin van het voorkomen van deze ziekte is niet mogelijk. Regelmatige controle van de symptomen en bijbehorende symptomen is belangrijk. Herhaalde controles van bloeddruk en nierfunctie, voedingsadviezen, fysiotherapie en ergotherapie, evenals logopedie zijn mogelijke therapeutische benaderingen.
Nazorg
In de meeste gevallen hebben degenen die lijden aan het Laurence-Moon-Biedl-Bardet-syndroom geen speciale follow-up-opties, dus een arts moet heel vroeg bij deze ziekte worden gecontacteerd en geraadpleegd. Zelfgenezing kan in de regel niet plaatsvinden, dus behandeling door een arts is altijd noodzakelijk.
Aangezien het Laurence-Moon-Biedl-Bardet-syndroom een erfelijke ziekte is, moet de betrokkene een genetisch onderzoek en advies laten uitvoeren als hij kinderen wil krijgen, zodat het Laurence-Moon-Biedl-Bardet-syndroom niet op zijn nakomelingen overgaat. wordt doorgegeven. In veel gevallen zijn de getroffenen afhankelijk van chirurgische ingrepen om de misvormingen en misvormingen te verlichten.
Hier moet de getroffen persoon na de procedure zeker rusten en voor zijn lichaam zorgen. In elk geval moeten inspanning of andere fysieke en stressvolle activiteiten worden vermeden om het lichaam niet onnodig te belasten. Omdat het Laurence-Moon-Biedl-Bardet-syndroom ook tot abnormaal gedrag kan leiden, moeten ouders het kind in ontwikkeling ondersteunen en aanmoedigen. Liefdevolle en intensieve gesprekken met het kind zijn ook nodig om psychische stoornissen of depressies te voorkomen.
U kunt dat zelf doen
Het Laurence-Moon-Biedl-Bardet-syndroom heeft verschillende symptomen, waarbij de patiënt vaak het meest lijdt aan een verminderde visuele functie. Zelfs bij kinderen begint het gebruikelijke zichtvermogen te verslechteren, zodat het de ouders zijn die het kind aan een arts aanbieden en zo de diagnose versnellen. Op deze manier kan de ziekte snel worden behandeld, hoewel de behandelingsopties tot nu toe alleen symptomatisch van aard waren.
De visuele stoornissen nemen bij de zieke kinderen steeds meer toe en schaden zo het dagelijks leven aanzienlijk, waardoor de kwaliteit van leven van de getroffenen afneemt. Omdat de problemen met het gezichtsvermogen voor de patiënt tal van moeilijkheden opleveren bij het naar school gaan, in hun vrije tijd en met betrekking tot hun fysieke integriteit. Ook de kans op ongevallen neemt aanzienlijk toe, bijvoorbeeld in het wegverkeer. Daarom begeleiden ouders waar mogelijk hun zieke kinderen of nemen ze verplegend personeel in, zodat de patiënt niet aan zijn lot wordt overgelaten.
In sommige gevallen verspreidt de ziekte zich tot het punt van blindheid. Omdat een dergelijke ontwikkeling op voorhand al duidelijk is, bereiden patiënten zich daarop voor. De ouders richten de leefruimte opnieuw in, zodat er geen gevaar voor slechtzienden in zit. Daarnaast leren de blinde slachtoffers hoe ze een lange stok moeten gebruiken, zodat ze zelfstandig buiten hun eigen huis kunnen bewegen.
.jpg)
.jpg)
.jpg)



.jpg)