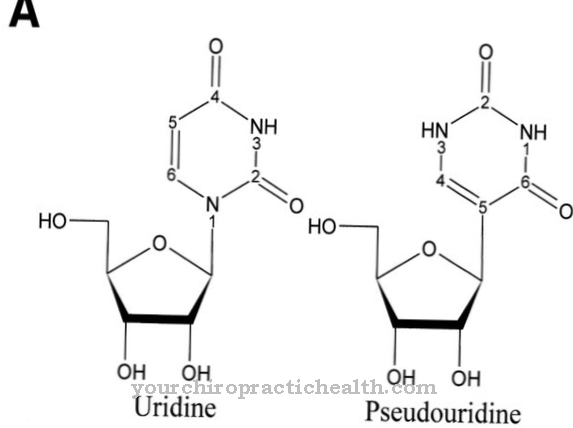
Onder de term Ciliopathie verschillende genetische ziekten worden samengevat, die een storing veroorzaken van de trilharen of de cellen die ze dragen.
Wat is ciliopathie?
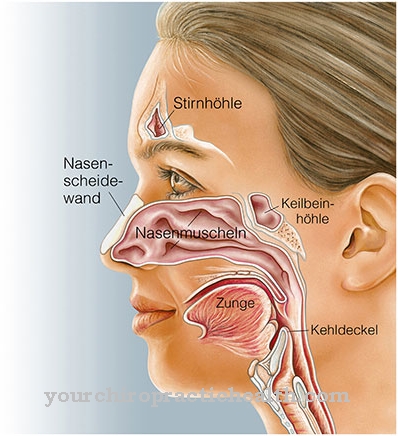
De cilia (ook wel cilia cilia genoemd) zijn cytoplasmatische uitsteeksels van de cel. Ze zijn tot 10 µm lang en tot 0,25 µm. De trilharen kunnen vrij bewegen en worden gebruikt om vloeistof- en slijmfilms te transporteren. De cilia-trilharen kloppen gelijkmatig achter elkaar, wat leidt tot een nog flikkerende stroom.
Dit zorgt ervoor dat de vloeistof of het slijm wordt afgevoerd. Bij de verschillende ziekten die worden genoemd Ciliopathieën worden gecombineerd, zijn de trilharen of de cellen die ze dragen beschadigd, waardoor ze hun taken niet meer in voldoende mate kunnen uitvoeren.
oorzaken
De ciliopathieën zijn genetische ziekten. Een groot aantal erfelijke ziekten kan nu worden toegewezen aan de ciliopathieën; voor sommigen is het definitieve bewijs nog niet geleverd. Ciliopathie is dus erfelijk.
De individuele ciliopathieën verschillen aanzienlijk van elkaar, omdat de trilharen zich in veel verschillende delen van het menselijk lichaam bevinden en daar verschillende taken uitvoeren. Als gevolg hiervan zijn de symptomen en diagnose van de ciliopathieën niet uniform, maar is er een aanzienlijk verschil tussen de individuele ziekten.
Symptomen, kwalen en tekenen
De symptomen, klachten en tekenen van een ciliopathie zijn niet uniform, aangezien de ciliopathie geen gesloten ziektebeeld is, maar gewoon een overkoepelende term voor verschillende erfelijke aandoeningen van de cilia. Om deze reden is een gestandaardiseerde diagnose niet mogelijk. De ciliopathieën omvatten een grote verscheidenheid aan ziekten met soms zeer verschillende symptomen:
- Kartagener-syndroom
- Meckel-Gruber-syndroom
- Joubert-syndroom
- Laurence Moon Biedl Bardet-syndroom (LMBBS)
- Nephronophthisis
- Senior Løken-syndroom
- Cyste lever
- Bardet-Biedel-syndroom
- Ellis van Creveld-syndroom (EVC)
- sommige soorten retinopathie
- bepaalde vormen van hydrocephalus
- Oro-facio-digitaal syndroom type 1 (OFD)
- recessieve en dominante erfelijke cystische nieren (ARPKD, ADPKD)
- Medullaire cystische nierziekte (ADMCKD)
- Alström-syndroom (ALMS)
- Short Rib Polydactyly Syndrome (SRPS)
- Cranio-ectodermale dysplasie
- verstikkende thoracale dysplasie (ATD)
Hieronder worden de symptomen en diagnose van het Kartagener-syndroom en het Laurence-Moon-Biedl-Bardet-syndroom (LMBBS) als voorbeelden gepresenteerd.
Het Kartagener-syndroom, ook bekend als primaire ciliaire dyskinesie, is een ciliopthia. Bij dit syndroom is er een disfunctie van de trilharencellen, vooral het respiratoire trilhaarepitheel. De beweging van de trilharen is verstoord, waardoor afscheidingen niet of onvoldoende kunnen worden verwijderd.
Hieruit volgt dat het zelfreinigende mechanisme van de bronchiën (mucociliaire klaring), dat wordt gedragen door het respiratoire trilhaarepitheel, ernstig verstoord is bij dit syndroom, wat te wijten is aan de disfunctie van de trilharen. Het Kartagener-syndroom wordt meestal overgeërfd als een autosomaal recessieve eigenschap.
Alle door trilharen bezette cellen van het lichaam worden aangetast, d.w.z. naast het respiratoire trilhaarepitheel worden ook de cellen van de oortrompet (gehoorbuis) en de neusbijholten aangetast. De focus van de symptomen ligt echter in de bronchiën, wat te wijten is aan het feit dat de meeste door trilharen bezette cellen daar worden aangetroffen.
Bij ongeveer 50 procent van de patiënten met het Katagener-syndroom treedt tijdens de embryonale fase een abnormale positie van de interne organen op in de vorm van een situs invertus, een spiegelbeeldige opstelling van de organen en bloedvaten. Getroffen mannen zijn meestal onvruchtbaar vanwege de verstoorde dyskinesie van het sperma. De meeste vrouwen zijn onvruchtbaar vanwege de dyskinesie van de trilhaartjes rond de eileider.
De meeste symptomen treden op in de luchtwegen. In de loop van de verstoorde zelfreiniging van de bronchiën treden vaak verstoppingen en infecties van de bronchiën op. De bronchitis is meestal recidiverend en moeilijk te behandelen. Bovendien zijn er frequente terugkerende en slecht behandelbare rhinitis, sinusitis en otitis media.
In de loop van de ziekte treedt vaak bronchiëctasie op, een onomkeerbare uitzetting van de bronchiën. Ademhalingsnoodsyndroom kan optreden bij pasgeborenen. Hydrocephalus, een uitbreiding van de binnenste of buitenste vloeistofruimten, kan ook voorkomen bij pasgeborenen.
Het Kartagener-syndroom is vaak merkbaar door een verhoogde vatbaarheid voor infecties bij kinderen. Als er naast een verhoogde vatbaarheid voor infectie een situs invertus is, kan het bestaan van het Kartagener-syndroom worden aangenomen. Als de situs invertus ontbreekt, is de diagnose moeilijk. Het bewijs van de aanwezigheid van het Kartagener-syndroom wordt geleverd door een elektronenmicroscopisch onderzoek van borsteluitstrijkjes of biopsieën van de relevante slijmvliezen.
Een causale therapie is niet mogelijk. De behandeling is symptomatisch. Met een vroege diagnose en adequate therapie kunnen de getroffenen een relatief normaal leven leiden.
Het Laurence-Moon-Bardet-Biedl-syndroom is ook een van de ciliopathieën. Het wordt gekenmerkt door een grote verscheidenheid aan mutaties en misvormingen die worden veroorzaakt door mutaties op verschillende chromosomen of genlocaties. De overerving is autosomaal recessief. Afhankelijk van de aangetaste genen kunnen verschillende symptomen optreden.
Deze zijn niet bij elke patiënt even uitgesproken. Deze symptomen zijn onder meer: zwaarlijvigheid; arteriële hypertensie; Suikerziekte; Korte gestalte; Hypotensie van de spieren; Misvormingen van de lever, eierstokken en galwegen; Hypogonadisme; Renale hypoplasie; Nierfalen; Pyelonefritis; motorische stoornissen; verstandelijke handicap; Retinitis pigmentosa; Blindheid; Anosmie; Gehoorverlies; Hemeralopia; een korte nek; merkbare hoeken van het ooglid; Polydactylie en syndactylie.
De diagnose van LMBBS is moeilijk omdat de symptomen ook voorkomen bij een groot aantal andere ziekten. Net als bij het Kartagener-syndroom wordt de uiteindelijke diagnose gesteld met behulp van een moleculair biologische test. Er is geen causale therapie; behandeling is symptomatisch.
Diagnose en ziekteverloop
De diagnose en het verloop van de ziekte zijn niet uniform voor de verschillende vormen van ciliopathieën. Zowel symptomen als diagnose en prognose verschilden aanzienlijk van elkaar. Wat de verschillende ciliopathieën gemeen hebben, is dat de diagnose kan worden bevestigd met moleculair biologische tests.
Complicaties
In de regel lijden mensen met ciliopathie aan een aantal verschillende syndromen en dus aan verschillende complicaties. Deze zijn ook sterk afhankelijk van de exacte ernst van de ziekte, zodat het meestal niet mogelijk is een algemene voorspelling te doen. In de eerste plaats leidt ciliopathie tot een infectie van de luchtwegen.
Dit leidt tot ademhalingsproblemen en mogelijk kortademigheid. De kwaliteit van leven van de getroffen persoon wordt aanzienlijk verminderd, zodat het niet langer mogelijk is om zonder meer zware activiteiten of sportieve activiteiten uit te voeren. De ontwikkeling van het kind wordt ook vertraagd door de ziekte.
In veel gevallen hebben de getroffenen ook heel vaak last van ontstekingen in de neus of luchtwegen. De meeste getroffenen hebben ook het vermogen zich niet voort te planten, waardoor ze ook last hebben van psychische klachten of depressies.
Er zijn geen bijzondere complicaties bij de behandeling van ciliopathie. Een volledige genezing is echter niet mogelijk, zodat de getroffenen in hun leven meestal altijd afhankelijk zijn van antibiotica en andere medicijnen.
Wanneer moet je naar de dokter gaan?
Als er binnen de familie een erfelijke ziekte wordt vastgesteld, moet samenwerking met een arts worden gezocht voordat een mogelijk kind wordt gepland. De potentiële ouders dienen zich vooraf uitgebreid te informeren over risico's of waarschijnlijke ontwikkelingen. Tijdens de zwangerschap moet u ook nauw samenwerken met een arts. De aangeboden preventieve onderzoeken zijn bedoeld om snel en volledig te kunnen reageren op mogelijke gezondheidsproblemen.
Omdat er geen causale therapieën kunnen worden gebruikt voor erfelijke ziekten, is vroege interventie bijzonder belangrijk. Als er geen kennis is van een familiegerelateerde genetische mutatie, worden afwijkingen vaak pas direct na de geboorte opgemerkt door leden van het verloskundig team. In een routinematig proces worden de nodige onderzoeken uitgevoerd om tot een diagnose te komen. Uiterlijk in de ontwikkeling van het kind zijn onregelmatigheden te zien in vergelijking met leeftijdsgenoten.
Raadpleeg een arts bij optische veranderingen, groeistoornissen of mentale afwijkingen. Storingen, eigenaardigheden van de reactie en onregelmatigheden in de bewegingssequenties moeten door een arts worden onderzocht. Ciliopathie is een overkoepelende term voor verschillende aandoeningen. Elk van hen vertoont individuele kenmerken bij de patiënt, zodat een controlebezoek bij een arts moet worden gestart als er een vermoeden bestaat van een bestaande gezondheidsverschillen.
Behandeling en therapie
Oorzakelijke therapieën zijn niet beschikbaar, de ziekten kunnen niet worden genezen, maar alleen verlicht door symptomatische therapie. De therapieën voor de verschillende ciliopathieën zijn ook verschillend.
preventie
Dit zijn genetische ziekten. Een preventie is dus niet mogelijk.
Nazorg
De nazorg voor ciliopathie is voornamelijk gebaseerd op het type en de ernst van de ziekte. Ziekten zoals het Laurence-Moon-Biedl-Bardet-syndroom of het Joubert-syndroom kunnen niet worden genezen. Nazorg zal zich richten op het beoordelen van behandelbare symptomen en het aanpassen van medicatie. Omdat de patiënten vaak chronisch ziek zijn, zijn er regelmatig vervolgonderzoeken.
Genezen klachten, zoals chronische pijn of vergiftigingsverschijnselen, dienen met medicijnen te worden behandeld. Als de ciliopathie gebaseerd is op een behandelbare aandoening, zoals een cystenier, hangt de nazorg af van het verloop van de ziekte en het succes van de therapie. Als de uitkomst positief is, kan de nier worden getransplanteerd.
Na zo'n niertransplantatie vindt wekelijks een onderzoek plaats in samenwerking met het transplantatiecentrum en de huisarts of specialist. De intervallen kunnen later worden teruggebracht tot vier keer per jaar. Onderdeel van de nazorg is de bepaling van bloedwaarden, radiologische onderzoeken zoals CT of MRI en andere onderzoeken afhankelijk van de onderliggende ziekte.
Tegelijkertijd wordt altijd de algemene toestand van de patiënt gecontroleerd. Als de ciliopathie het gevolg is van andere ziekten, moet de betreffende specialist worden geraadpleegd. In ieder geval maken een specialist in nierziekten en de huisarts deel uit van het medisch team.
U kunt dat zelf doen
Ciliopathieën kunnen een grote verscheidenheid aan vormen aannemen en moeten altijd afzonderlijk worden behandeld. Algemene maatregelen die het herstel kunnen bevorderen, zijn fysiotherapie en een verandering in levensstijl. Vormen zoals het Joubert-syndroom of het Laurence-Moon-Bardet-Biedl-syndroom worden altijd symptomatisch behandeld, waarbij de patiënt gezond moet eten, voldoende moet bewegen, maar voor zichzelf moet zorgen. Dit verlicht in ieder geval de symptomen.
Als medicatie is voorgeschreven, moet zorgvuldig worden gelet op eventuele bijwerkingen en interacties, aangezien deze een negatief effect kunnen hebben op de ontwikkeling van andere ciliopathieën. Als zich complicaties voordoen, moet de arts hierover worden geïnformeerd. Patiënten dienen een klachtendagboek bij te houden en alle waarneembare symptomen en klachten gedetailleerd op te schrijven.
De verantwoordelijke arts moet in elk geval beslissen welke maatregelen nuttig zijn bij ciliopathie. Door de verschillende vormen van lijden is een individueel behandelplan altijd noodzakelijk. Getroffenen kunnen het beste contact opnemen met hun huisarts, die verdere tips kan geven over hoe de medische behandeling het beste kan worden ondersteund. Verder kan hij contact leggen met zelfhulpgroepen die de patiënt van verdere maatregelen kunnen voorzien.

.jpg)
.jpg)
.jpg)

.jpg)

.jpg)
.jpg)


.jpg)