De Ik celziekte is een lyosomale mucolipidose. De oorzaak van de stapelingsziekte is een mutatie van het GNPTA-gen met het q23.3-genlocus op chromosoom 12. Symptomatische behandeling wordt voornamelijk uitgevoerd door het toedienen van bisfosfonaten.
Wat is een celziekte?

© red150770 - stock.adobe.com
Stapelingsziekten worden gekenmerkt door de afzetting van verschillende stoffen in cellen en organen van het menselijk lichaam. Het is een heterogene groep ziekten die kan worden onderverdeeld in verschillende subvormen. Naast glycogenosen, mucopolysaccharidosen en lipidosen, onderscheidt de geneeskunde zich, afhankelijk van de afgezette stof, van sfingolipidosen, hemosiderosen en amyloïdoses.
Lysosomale stapelingsziekten tasten de lysosomen aan. Dit zijn kleine, met membraan beklede celorganellen in de eukaryoten. Lysosomen worden gevormd door het Golgi-apparaat en zijn uitgerust met hydrolytische enzymen en fosfatasen. Met behulp van hun enzymen moeten ze voornamelijk vreemde stoffen en lichaamseigen stoffen verteren.
I-celziekte is een lyosomale mucolipidose met twee verschillende subtypes. Leroy en DeMars documenteerden de ziekte voor het eerst in de jaren zestig en wezen op de gelijkenis ervan met mucopolysaccharidose type I, bekend als de ziekte van Hurler. De naam van de ziekte komt van de fibroblastinsluitsels, de zogenaamde inclusiecellen, in de huid van de patiënt.
oorzaken
De oorzaak van de I-celziekte ligt in een gebrek aan activiteit van het N-acetylglucosaminyl-1-fosfotransferase. De beperkte activiteit van dit enzym verhindert dat een groot deel van de lysosomale enzymen het inwendige van het lysosoom binnendringt. De regulatie van de lysosomale enzymen wordt bepaald door de activiteit van de fosfotransferase.
Het maakt de synthese van een sorteersignaal in een gezond organisme mogelijk. Dit proces is verstoord bij I-celziekte. Daarom is er geen labeling met mannose-6-fosfaat. Om deze reden zijn de lysosomale enzymen niet meer goed gesorteerd en migreren ze ongecontroleerd via het plasmamembraan in de extracellulaire matrix.
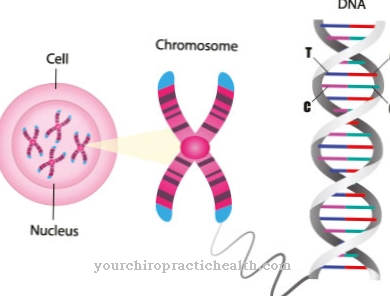
De reden hiervoor is een mutatie in het GNPTAB-gen. Het verwijdert de functionaliteit van het N-acetylglucosaminyl-1-fosfotransferase en daarmee het vermogen om de synthese van mannose-6-fosfaat te katalyseren. Het transport van lysomale enzymen is zo verstoord. Het N-acetyl-glucosamine-1-fosfotransferase bestaat uit de subeenheden alfa, bèta en gamma. Ze zijn gecodeerd op twee genen.
De erfelijke I-celziekte beïnvloedt het GNPTA-gen op chromosoom 12. Er is een mutatie in de q23.3-genlocus. Voor de zeldzame ziekte wordt een incidentie van ongeveer 0,3: 100.000 gegeven. Overerving is onderhevig aan autosomaal recessieve overerving. Beide ouders moeten dus het defecte gen dragen om de ziekte door te geven.
Symptomen, kwalen en tekenen
In de meeste gevallen kunnen de symptomen van I-celziekte onmiddellijk na de geboorte of uiterlijk een paar maanden later worden waargenomen, en hun kenmerken zijn vergelijkbaar met die van het Hurler-syndroom. In tegenstelling tot patiënten met het Hurler-syndroom vertonen patiënten met I-celziekte geen uitscheiding van mucopolysacchariden.
De individuele symptomen van de ziekte zijn onderhevig aan een groot aantal variaties. Kornfeld en Sly vatten klinische kenmerken van het skelet, de inwendige organen, de ogen, de huid, het centrale zenuwstelsel en het gezicht samen. Het skelet wordt zo vaak aangetast door kyfoscoliose en heupdislocaties.
Clubvoeten, gewrichtscontracturen en misvormingen van de wervels kunnen ook aanwezig zijn. Hetzelfde geldt voor een kleine gestalte en dysostosemultiplex. De ziekte kan zich in de inwendige organen manifesteren in de vorm van hepatosplenomegalie en cardiomegalie of hartaandoeningen. Het gezicht van de patiënt heeft grover gelaatstrekken.
Exophthalmus, hyperplastisch tandvlees of scaphocefalie zijn typische symptomen. Kenmerkend zijn ook een open mond en een diep ingevallen neus. De ogen van de getroffenen hebben vaak troebelheid van het hoornvlies of gezwollen oogleden. De huid is dik en grof, met ernstige psychomotorische of mentale retardatie in het centrale zenuwstelsel.
Diagnose en ziekteverloop
De eerste vermoedelijke diagnose van I-celziekte kan worden gesteld door visuele diagnose op basis van de anamnese. Een biochemische bepaling van de lysosomale enzymactiviteit in het serum kan worden gebruikt om de diagnose te bevestigen. Deze bepaling onthult een absurde relatie tussen intra- en extracellulaire activiteit.
De activiteit van fosfotransferase in de fibroblasten kan ook worden bepaald om de diagnose te bevestigen. De insluitsels komen overeen met mucopolysachariden, lipiden of oligosachariden. Moleculair genetische diagnostiek kan alle resterende twijfels wegnemen. Als er een passende geschiedenis is, kan de ziekte ook worden gediagnosticeerd als onderdeel van de prenatale diagnose.
Vanwege de lage prevalentie is prenatale work-up eigenlijk alleen aan te raden als er sprake is van een gezinssituatie. Het verloop van de ziekte hangt af van de symptomen in het individuele geval en is niet direct voorspelbaar. De meeste patiënten overleven de leeftijd van tien jaar echter nauwelijks. Mildere vormen van ontwikkeling zijn echter in individuele gevallen niet geheel uitgesloten.
Complicaties
I-celziekte kan leiden tot verschillende complicaties en klachten. Deze worden echter laat herkend, zodat de I-celziekte pas laat kan worden vastgesteld. De symptomen zijn relatief inconsistent, wat de behandeling vaak bemoeilijkt. Dit leidt meestal tot ongemak en misvormingen van de huid, ogen en inwendige organen.
In het ergste geval kan de getroffen persoon blind worden of direct overlijden aan orgaanfalen. Verder is er een uitgesproken kleine gestalte en ook hartproblemen. De oogleden zijn vaak opgezwollen en er is sprake van verminderde intelligentie en mentale achterstand. Het is niet ongebruikelijk dat de getroffen persoon in het dagelijks leven afhankelijk is van de hulp van andere mensen vanwege de vertraging om ermee om te gaan.
De kwaliteit van leven van de patiënt wordt sterk verminderd door de I-celziekte. In de regel zijn er geen bijzondere complicaties bij de behandeling van de ziekte. Er worden medicijnen en psychologische behandelingen gebruikt die de symptomen kunnen verlichten. Een volledige en oorzakelijke behandeling van deze ziekte is echter niet mogelijk. De levensverwachting wordt verminderd door de ziekte.
Wanneer moet je naar de dokter gaan?
I-celziekte wordt meestal onmiddellijk na de geboorte van het kind gediagnosticeerd. Of verdere behandelingsmaatregelen nodig zijn, hangt af van het type en de ernst van de symptomen. Kleine misvormingen hoeven niet noodzakelijk te worden behandeld. Clubvoeten en misvormingen van de wervels daarentegen zijn ernstige misvormingen die operatief en met medicatie moeten worden behandeld. Ouders dienen onmiddellijk een specialist te raadplegen als de verantwoordelijke arts van het kraamkliniek dat nog niet heeft gedaan.
Als er naar aanleiding van de klachten een ongeval of val optreedt, moet het kind naar het ziekenhuis worden gebracht of moeten de ouders direct de hulpdiensten bellen. Bij ernstige misvormingen, die op latere leeftijd ook de psyche van het kind kunnen aantasten, moet een therapeut worden geraadpleegd om de medische behandeling te begeleiden. De I-celziekte vereist daarom altijd een medisch onderzoek. De juiste contactpersoon is de kinderarts of een specialist in erfelijke ziekten. Bij visuele stoornissen dient een oogarts te worden geraadpleegd.
Artsen & therapeuten bij u in de buurt
Behandeling en therapie
De I-celziekte wordt als ongeneeslijk beschouwd. Een causale therapie bestaat dus niet. De behandeling is alleen symptomatisch en ondersteunend. Psychotherapeutische zorg voor getroffen gezinnen maakt een groot deel uit van ondersteunende therapie. Symptomatische therapie is afhankelijk van het individuele geval. De botklachten worden vaak behandeld door bisfosfonaten te geven.
Deze medicijnen zijn bekend van de behandeling van osteoporose en hebben een hoge affiniteit voor het botoppervlak. Vooral in het gebied van de resorptielacunes hechten ze zich aan de botten. Ze remmen daarbij de botafbrekende osteoclasten en verminderen zo de botresorptie. De medicijnen zijn pyrofosfaatanalogen met een koolstofhoudende P-O-P-binding.
Er vindt geen enzymatische hydrolyse op plaats. De aminobisfosfonaten behoren tot de meest recente van deze stoffen. Bovendien zijn alendronaat, clodronaat, etidronaat, ibandronaat, pamidronaat en risendronaat in Duitsland goedgekeurd uit dezelfde geneesmiddelengroep. Hetzelfde geldt voor tiludronaat en zoledronaat.
Naast deze medicijnen kunnen beenmergtransplantaties ook worden gebruikt om I-celziekte te behandelen. Het succes van deze behandeling was slechts beperkt in eerdere gevallen. Gentherapieën worden nu onderzocht als een nieuwe therapeutische benadering voor gendefecten. Gentherapieën hebben aanvankelijk succes getoond in diermodellen. Tot dusverre konden ze in de praktijk niet op mensen worden gebruikt. Deze relatie zal in de toekomst waarschijnlijk veranderen.
Uw medicatie vindt u hier
➔ Geneesmiddelen tegen pijnOutlook & prognose
I-celziekte is een erfelijke ziekte die nog niet symptomatisch is behandeld. De prognose is dienovereenkomstig negatief. Hoewel de symptomen aanzienlijk kunnen worden verminderd door vroege therapie, neemt de I-celziekte bijna altijd een ernstig beloop.
De kleine gestalte en de beschadiging van de inwendige organen en het hoofd verkorten de levensverwachting al aanzienlijk. Bovendien kunnen misvormingen in het gezicht, de huid en de ogen de levensverwachting verminderen, maar vooral ook de kwaliteit van leven van de getroffen persoon. Sommige van de getroffenen bereiken de leeftijd van 40 of 50 jaar, maar de meesten van hen sterven in de kindertijd of adolescentie.
Als de I-celziekte niet wordt behandeld, sterven de zieken vaak in de eerste levensjaren. De prognose is daarom nogal negatief. Desalniettemin wordt uitzicht op een relatief symptoomvrij leven geboden als de patiënt wordt behandeld als onderdeel van een integrale therapie en, indien nodig, wordt geplaatst in een instelling voor lichamelijk gehandicapten. Fysiotherapie en therapeutische maatregelen kunnen het welzijn van de patiënt op lange termijn aanzienlijk verbeteren.
preventie
I-celziekte kan alleen worden voorkomen door een moleculair genetische test voorafgaand aan gezinsplanning. Als onderdeel van de prenatale diagnose kunnen aanstaande ouders ook besluiten de zwangerschap af te breken.
Nazorg
In de meeste gevallen hebben degenen die lijden aan I-celziekte geen of zeer weinig vervolgmaatregelen beschikbaar. De ziekte moet zo vroeg mogelijk door een arts worden herkend, zodat een verdere verergering van de symptomen kan worden voorkomen. Aangezien dit een genetisch bepaalde ziekte is, moet bij een kinderwens altijd eerst een genetisch onderzoek en consult worden uitgevoerd om te voorkomen dat de nageslacht wordt overgeërfd van de I-celziekte.
De meeste patiënten zijn voor deze ziekte afhankelijk van verschillende medicijnen. Het is belangrijk om ervoor te zorgen dat de dosering correct is en dat de medicatie regelmatig wordt ingenomen. Als iets onduidelijk is, er zijn bijwerkingen of als u vragen heeft, dient u altijd eerst een arts te raadplegen.
Evenzo hebben veel patiënten psychologische ondersteuning nodig bij deze ziekte, hoewel liefdevolle gesprekken met ouders of familieleden een positief effect kunnen hebben op het beloop van de ziekte. Een getroffen persoon heeft in het dagelijks leven de hulp en steun van zijn eigen familie nodig. In veel gevallen beperkt of verkort de I-celziekte de levensverwachting van de getroffen persoon aanzienlijk.
U kunt dat zelf doen
Patiënten die lijden aan I-celziekte kunnen hun toevlucht nemen tot verschillende conservatieve en alternatieve behandelmethoden. Conservatieve therapie richt zich op het verlichten van symptomen en kwalen.
Het gebruik van hulpmiddelen zoals krukken of orthopedische inlegzolen kan het verloop van de betreffende misvormingen vertragen en zo ook de pijn verminderen. Medicatie helpt pijn te verlichten en kan worden aangevuld met alternatieve maatregelen zoals massage of acupunctuur. Alternatieve therapieën dienen vooraf met de verantwoordelijke arts te worden besproken. De arts kan de patiënt mogelijk rechtstreeks doorverwijzen naar een homeopaat of verdere tips geven over hoe het betreffende symptoom moet worden behandeld.
Aangezien de I-celziekte ondanks alle behandelingsopties meestal dodelijk is, moet therapeutisch advies worden ingewonnen. Niet alleen de getroffenen zouden hun angsten moeten overwinnen. De familieleden en vrienden hebben meestal ook ondersteuning nodig bij het omgaan met de ziekte en de mogelijke negatieve afloop ervan. Deelname aan een zelfhulpgroep is ook een optie voor de patiënt en diens naasten. Contact met andere patiënten helpt de ziekte te aanvaarden, en vaak kunnen andere patiënten ook verdere behandelingsmaatregelen en strategieën voorstellen voor het dagelijks leven met I-celziekte.
.jpg)







.jpg)

.jpg)
.jpg)
