Er zijn in totaal 45 verschillende lysosomale stapelingsziekten, die een heterogene groep van aangeboren stofwisselingsziekten vormen. Mensen die aan een van deze ziekten lijden, hebben een genetisch defect. Alle stapelingsziekten hebben één ding gemeen: een bepaald enzym is afwezig of slechts gedeeltelijk functioneel.
Wat is lysosomale stapelingsziekte?

© designua - stock.adobe.com
Deze aangeboren stapelingsziekten zijn zeldzaam, aangezien minder dan vijf op de 10.000 mensen er last van hebben. De verschillende ziekten hebben een heel verschillend verloop en de symptomen kunnen sterk variëren.
De meest bekende vormen van lysosomale stapelingsziekte zijn de ziekte van Fabry, de ziekte van Gaucher, de ziekte van Pompe en mucopolysaccharidose (MPS). Ze worden vaak "weeskinderen" genoemd omdat de weg naar een specifieke diagnose en geschikte therapie erg lang kan zijn. Soms kan het jaren duren voordat de getroffenen erachter komen wat er met hen gebeurt.
oorzaken
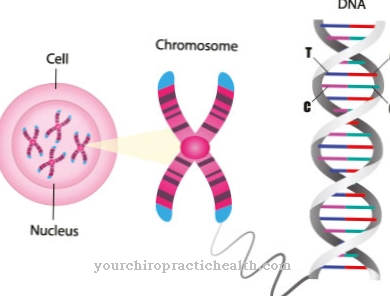
Lysosomale stapelingsziekten worden gekenmerkt door bepaalde vormen van erfelijke stofwisselingsziekten. De patiënt mist een belangrijk enzym dat ervoor zorgt dat het metabolische evenwicht goed verloopt. In de minder uitgesproken vorm is dit enzym in ieder geval niet in voldoende mate aanwezig.
De taak van enzymen is om via de stofwisseling via de lysosomen de vervuilende stoffen en afvalstoffen die zich in het menselijk organisme ophopen af te voeren of weer zo te verwerken dat er geen symptomen optreden.
Als er een enzymtekort is, is deze soepel werkende verwijderingscyclus niet langer gegarandeerd. De schadelijke stoffen nestelen zich in de cellen en verstoren de stofwisselingscyclus. In de beginfase hebben de verstoringen geen merkbaar effect, er zijn slechts enkele beperkingen. Als deze stofwisselingsstoornis echter onbehandeld blijft als gevolg van een enzymdeficiëntie, vermenigvuldigen de symptomen zich doordat de cellen erg vergroot worden.
Symptomen, kwalen en tekenen
In het ergste geval gaan deze onder. De gevolgen zijn schade aan de botten, het zenuwstelsel, milt, nieren, spieren of hart. Door de verminderde of afwezige enzymactiviteit zorgt de ziekte van Fabry ervoor dat vet (globotriaosylceramide, Gb3) in de cellen wordt opgeslagen. Deze ongewenste afzettingen kunnen leiden tot ernstige pijn in de tenen of vingers, beroerte en nierbeschadiging.
Diagnose en ziekteverloop
Dit ziektebeeld beïnvloedt verschillende systemen tegelijkertijd: bloedvaten, nieren, hart en zenuwstelsel. De autosomaal recessieve ziekte van Gaucher veroorzaakt een mutatie van het enzym ‘beta-glucocerebrosidase’ en leidt tot een ophoping van substraat in de cellen, vooral in de macrofagen (fagocyten) die tot het reticulo-endotheliale systeem behoren. Het aantal bloedcellen verandert, de lever en de milt worden vergroot en de botten doen pijn.
De ziekte is progressief en meestal etnisch, aangezien het in de meeste gevallen voorkomt bij mensen van joodse afkomst. De ziekte van Pompe wordt ook wel "zuur-maltase-deficiëntie" genoemd. Het ziektebeeld behoort tot de groep van glycogenese type II De getroffen personen missen het enzym "alpha-1,4-glucosidase" (zure maltase) of het is niet in voldoende hoeveelheden beschikbaar. Door de verstoorde glycogeenafbraak in de spieren hebben patiënten last van de vernietiging van de spiercellen in de vorm van suikeropslag.
Mucopolysaccharidose type I (MPS), ook wel bekend als de ziekte van Hunter, heeft verschillende klinische oorzaken. De ziekte van Hurler is de meest ernstige vorm en de ziekte van Scheie bevindt zich aan het einde van de klinische pathogenese. Er zijn overgangen met verschillende kenmerken tussen deze twee vormen van progressie. De meest opvallende eigenschap is de verminderde afbraak van koolhydraten die zich ophopen in de lysosomen van de cellen.
Patiënten met de ziekte van Hunter kunnen een kleine gestalte, vergrote milt en lever, grove kenmerken, verdikte huid, vergrote tong en ademhalingsmoeilijkheden ervaren. Daarnaast wordt het skelet vaak veranderd in het gebied van het bekken, de wervelkolom, de handbeenderen en de schedel. Navelstreng en [[liesbreuken] zijn mogelijk.
Complicaties
In de meeste gevallen treden symptomen of complicaties erg laat op bij deze ziekte. Hierdoor wordt de diagnose laat gesteld, waardoor vroege behandeling in de meeste gevallen onmogelijk is. Zonder behandeling treden naarmate de ziekte voortschrijdt diverse klachten en schade aan de inwendige organen op.
Vooral de nieren, lever en milt worden aangetast. Het hart kan ook door deze ziekte worden aangetast, wat in het ergste geval kan leiden tot hartdood. Bovendien treedt schade aan de nieren op en hebben getroffenen vaak last van pijn in de tenen of vingers. Verlamming kan ook optreden als de hersenen door deze ziekte zijn beschadigd. De lever en milt kunnen vergroot zijn en ook hevige pijn veroorzaken.
Het is niet ongebruikelijk dat de botten van de aangedane persoon broos en ook pijnlijk zijn. Behandeling van deze ziekte blijkt moeilijk. In veel gevallen is de levensverwachting van de getroffen persoon aanzienlijk verminderd. Er zijn meestal geen bijzondere complicaties bij de behandeling met medicatie. Een positief verloop van de ziekte kan echter niet in alle gevallen worden gegarandeerd.
Uw medicatie vindt u hier
➔ Geneesmiddelen tegen pijnWanneer moet je naar de dokter gaan?
Haaruitval, gewrichtsproblemen en orgaanaandoeningen zijn mogelijke tekenen van lysosomale stapelingsziekte. Een bezoek aan de dokter is aan te raden als de symptomen steeds terugkeren of plotseling optreden zonder dat er een oorzaak wordt gevonden. Als de symptomen verband houden met een eerder gediagnosticeerd enzymdefect of een andere ernstige ziekte, moet de verantwoordelijke arts worden geraadpleegd. Een onbehandelde stapelingsziekte kan leiden tot dementie, onvruchtbaarheid, neuropathieën en andere complicaties, waarvan sommige levensbedreigend zijn. Daarom moeten alle denkbare symptomen worden onderzocht, zelfs als er geen specifiek vermoeden is.
De symptomen van lysosomale stapelingsziekte kunnen in fasen optreden of zich verraderlijk ontwikkelen, maar vereisen altijd onderzoek en behandeling. Getroffen mensen kunnen het beste rechtstreeks met hun huisarts of een internist praten. De eigenlijke therapie vindt meestal plaats in een gespecialiseerde kliniek voor inwendige ziekten, hoewel fysiotherapie of psychotherapie afhankelijk van de symptomen kan worden aangesloten. In het bijzonder zijn therapeutische maatregelen aangewezen vanwege het vaak negatieve verloop van de ziekte.
Therapie en behandeling
Afhankelijk van hoe vroeg een adequate diagnose wordt gesteld, kunnen deze erfelijke ziekten heel goed worden behandeld met enzymvervangende therapie, waardoor de getroffenen veel minder klachten hebben en dus een betere kwaliteit van leven. Deze vervangingstherapie wordt toegepast volgens het ziektebeeld.
Mensen die aan de ziekte van Gaucher lijden, missen het "enzym ß-glucocerebrosidase", dat biotechnologisch wordt geproduceerd en in het lichaam van de patiënt wordt ingebracht. Lysosomen werken efficiënt en zijn in staat stoffen uit hun directe omgeving op te nemen. Om deze reden worden de kunstmatig gebruikte enzymen zodanig gemodificeerd dat ze op een ideale manier aan de lysosomen kunnen worden toegevoerd.
De macrofagen (fagocyten) breken de glucocerebrosiden af die zich in de cellen hebben opgehoopt. Deze therapie is te vergelijken met insulinetherapie bij diabetes mellitus, met het verschil dat het geen ontbrekend hormoon is maar een enzym dat niet wordt aangevoerd. Het lichaam breekt regelmatig alle stoffen af, inclusief het aangeleverde kunstmatige enzym.
Vanwege deze regelmatige afbraak van de stof moeten patiënten deze infusietherapie tot het einde van hun leven regelmatig ondergaan. De enzymvervangingstherapie werkt niet symptomatisch, maar bestrijdt direct de oorzaak van de erfelijke ziekte. Artsen noemen deze therapie causaal. De principes van therapie zijn van toepassing op alle vier de hierboven genoemde veel voorkomende stapelingsziekten.
Pompe-patiënten worden ook behandeld met infusietherapie. Bij deze ziekte wordt het niet-bestaande enzym "zuur alfa glucosidase" geleverd en helpt het glycogeen af te breken dat zich heeft opgehoopt in de lysosomen van de spieren. Bij patiënten met de ziekte van type "mucopolysaccharidose type I" is het lysosomale enzym "alpha-iduronidase" niet of niet in voldoende hoeveelheden aanwezig. Het is een van de zeldzaamste stapelingsziekten waarbij suikermoleculen zich ophopen in organen en weefsels.
Als het proces normaal is, breekt het enzym mucopolysacchariden af. De suikermoleculen zijn langgeketend en zijn betrokken bij de ontwikkeling van steun- en bindweefsel, bijvoorbeeld botten, huid, gewrichtsvloeistoffen en kraakbeen. Als het normale verloop van de afbraak wordt verstoord door een gebrek aan enzym, hopen pathologische glycosaminoglycanen (GAG) zich op in de individuele cellen. Toekomstige therapie-opties zijn gericht op het innemen van tabletten.
Outlook & prognose
De prognose voor stapelingsziekte is slecht. Een genetische aanleg bleek de oorzaak van de gezondheidsstoornis te zijn. Wettelijke vereisten verbieden artsen en wetenschappers om de menselijke genetica te veranderen. Om deze reden blijft de ziekte levenslang en heeft geen uitzicht op herstel.
De behandelende arts concentreert zich op het behandelen van de symptomen die optreden. Indien onbehandeld, zullen verschillende klachten in de loop van de tijd toenemen. Het botstelsel is beschadigd en er ontstaan problemen aan de organen. In het ergste geval zullen de interne organen defect raken en uiteindelijk zal hun functie mislukken. Hierdoor dreigt de betrokkene met vroegtijdig overlijden.
De uitdaging van de ziekte ligt in de diagnose. Opvallende en sterk waarneembare klachten treden bij een groot aantal patiënten pas op latere leeftijd op. Hierdoor blijft de erfelijke aandoening lange tijd onopgemerkt en is vroege behandeling van de ziekte moeilijk. Hoe later een diagnose wordt gesteld, hoe ongunstiger het verloop is. In een vergevorderd stadium van de ziekte zijn de inwendige organen of gewrichten al ernstig beschadigd. Chirurgische ingrepen zijn vereist en als de ziekte ongunstig verloopt, kan slechts één donororgaan het leven van de getroffen persoon redden. Vroegtijdige behandeling is daarom essentieel voor een betere prognose.
preventie
Omdat het een aangeboren genetisch defect is dat de expressie van een enzym verhindert, kan deze ziekte niet preventief worden behandeld. De nieuwste resultaten op het gebied van genetische manipulatie zouden echter een benadering op dit gebied kunnen bieden.
Nazorg
Bij deze ziekte lijden mensen aan een aantal verschillende complicaties en aandoeningen. Deze hebben in de regel allemaal een zeer negatief effect op de kwaliteit van leven van de getroffen persoon, zodat de diagnose al heel vroeg moet worden gesteld. Hoe eerder een arts wordt geraadpleegd, hoe beter het verloop van deze ziekte gewoonlijk is.
De ernst van deze ziekte kan sterk variëren, waardoor een algemene voorspelling vaak niet mogelijk is. De getroffenen lijden aan ernstige schade aan de interne organen. De nieren en het hart worden primair aangetast, zodat het kind de eerste dagen kan overlijden als de symptomen niet op tijd worden verholpen. Er zijn ook vetophopingen in verschillende delen van het lichaam.
Vooral de vingers en tenen worden aangetast, wat kan leiden tot een aanzienlijk verminderde esthetiek voor de getroffen persoon. In de regel treedt in het verdere verloop schade aan de nieren en de hersenen op, zodat de getroffen persoon aan deze schade overlijdt. De ouders en familieleden lijden ook vaak aan depressies of andere psychische stoornissen als gevolg van de ziekte.
U kunt dat zelf doen
Lysosomale stapelingsziekten vereisen vaak intensieve medische zorg. Vaak zijn er niet genoeg mogelijkheden voor zelfhulp. De ouders van getroffen kinderen ervaren vaak ernstige stress in hun thuisomgeving omdat hun kind constante zorg en aandacht nodig heeft.
De ziektebeelden van de individuele stapelingsziekten zijn verschillend. Er zijn zowel gemakkelijke als zeer moeilijke vormen. Een voorbeeld is de ziekte van Gaucher. De hulp van ouders beperkt zich vaak tot het voeden van het ernstig gehandicapte kind. In mildere gevallen kan de levensverwachting bijna normaal zijn. Niettemin is constant medisch toezicht noodzakelijk om mogelijke complicaties te voorkomen. Regelmatige lichamelijke activiteit is een van de begeleidende therapieën die ook thuis uitgevoerd kunnen worden. Verder moet er een zorgvuldig kankerscreeningonderzoek komen. Dit vereist constante bezoeken aan de dokter met hun kind van de ouders. Hetzelfde geldt voor andere lysosomale stapelingsziekten.
Bij sommige ziekten kunnen naast lichamelijke handicaps ook psychische stoornissen optreden, die toch bijzondere ondersteuning behoeven. Bij mildere vormen van bepaalde ziekten, zoals de ziekte van Hunter, treden aanvankelijk alleen skeletveranderingen en aangezichtsdysmorfisme op. Hier kan de getroffen patiënt echter vaak een zelfstandig leven leiden. Hier zijn echter ook constante medische onderzoeken nodig om mogelijke complicaties zoals hartfalen of aandoeningen van de luchtwegen uit te sluiten. De patiënt kan omgaan met psychische stress veroorzaakt door lichamelijke vervormingen door middel van psychologische begeleiding.
.jpg)

.jpg)


.jpg)