Smith-Magenis-syndroom is de naam voor een zelden voorkomende erfelijke ziekte. Het wordt veroorzaakt door een verlies van chromosoom 17.
Wat is het Smith-Magenis-syndroom?

© vchalup– stock.adobe.com
Bij de Smith-Magenis-syndroom (sms) is een zeldzame genetische ziekte. De aangedane persoon mist een klein stukje chromosoom 17. Dit kleine deel is echter belangrijk omdat het informatie bevat die belangrijk is voor de gezonde rijping van het kind in de baarmoeder.
De twee genetica-specialisten Ann Smith en Ellen Magenis zijn de naamgenoot van het Smith-Magenis-syndroom. Begin jaren tachtig beschreven de doktoren de ziekte voor het eerst. Volgens Amerikaanse studies is de prevalentie van het Smith-Magenis-syndroom 1 op 25.000 geboorten. Tot nu toe is de ziekte slechts sporadisch in Duitstalige landen voorgekomen. Er zijn geen verschillen tussen de geslachten bij het optreden van het syndroom.
oorzaken
Het Smith-Magenis-syndroom wordt veroorzaakt door een gebrek aan een klein deel van chromosoom 17, wat betekent dat de informatie daar ook ontbreekt. In de geneeskunde wordt het ontbreken van deze gegevens aangeduid als schrapping 17p11.2. De p staat voor "petit", wat "klein" betekent en duidt op de korte chromosoomarm.
Hoeveel van het chromosoomstuk ontbreekt, verschilt van persoon tot persoon. Om deze reden varieert de intensiteit van het Smith-Magenis-syndroom. Er zijn verschillende genen in het ontbrekende deel van het chromosoom. De symptomen van het Smith-Magenis-syndroom worden echter voornamelijk veroorzaakt door het ontbreken van het RAI1-gen.
Sommige medische professionals suggereren dat het ontbreken van de andere genen bijdraagt aan de individuele verschillen in ziektekenmerken. Er wordt nog steeds wetenschappelijk onderzoek gedaan naar de exacte relaties. Volgens de huidige wetenschappelijke kennis zijn er geen risicofactoren zoals omgevingsinvloeden of wangedrag van de moeder tijdens de zwangerschap.
Dit is hoe het Smith-Magenis-syndroom zich spontaan ontwikkelt. De mutatie op het 17e chromosoom wordt door toeval veroorzaakt en begint vóór de bevruchting. Mutatie is mogelijk bij zowel de moeder als de vader. Het kan echter gebeuren dat de kinderen van getroffen ouders ondanks de deletie op chromosoom 17 gezond blijven en niet automatisch ziek worden.
Symptomen, kwalen en tekenen
Het Smith-Magenis-syndroom gaat gepaard met een aantal typische gedragsproblemen. Deze omvatten ontwikkelingsstoornissen zoals matige tot ernstige mentale ontwikkelingsstoornissen. Dit zijn meestal vertragingen in de taalontwikkeling en algemene ontwikkelingsachterstand.
Aangezien het Smith-Magenis-syndroom een dysmorf syndroom is, zijn er ook dysmorfologische afwijkingen zoals brachycefalie (korthoofdigheid), hypoplasie van het middengezicht, brede en korte handen, diepliggende ogen, gebogen bovenlip, mondhoeken die naar beneden wijzen, diepliggende oren en brede oren Wortel van de neus.
Verder zijn er gedragsproblemen die bij kleine kinderen niet altijd voldoende herkend kunnen worden. De getroffenen lopen zelfverwondingen op door met hun hoofd tegen de muur te slaan, in hun vingers of handen te bijten en teennagels of vingernagels uit te trekken.
De pijn wordt als verminderd gevoeld. Omdat ook het dag-nachtritme verstoord is, hebben de patiënten last van uitgesproken slaapstoornissen. Daarnaast zijn er gezondheidsproblemen zoals perceptief of conductief gehoorverlies, kleine gestalte of scoliose.
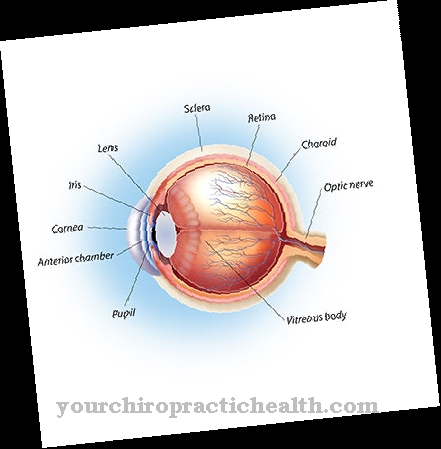
Verder zijn er veranderingen in de ogen zoals bijziendheid, strabismus of een te klein hoornvlies, een hartafwijking, epilepsie, hersenafwijkingen zoals ventriculomegalie of urineblaasklachten.
Kenmerkend zijn ook een diepe en ruwe stem, vermoeidheid overdag, lage schildklierhormoonspiegels en een verlaagd immunoglobulineniveau. Bovendien gedragen veel patiënten zich als autistische mensen, zijn ze niet in staat om te praten, streven ze een uniform, routineus leven na en zijn ze bang om aangeraakt te worden. Het is niet ongebruikelijk dat agressief gedrag zelfs uitbarstingen van woede kan veroorzaken.
Diagnose en ziekteverloop
Omdat de symptomen van het Smith-Magenis-syndroom extreem uitgebreid zijn, zijn ze niet voldoende voor een exacte diagnose van de ziekte. Er zijn enkele ziekten die lijken op het Smith-Magenis-syndroom. Deze omvatten het Prader-Willi-syndroom. Het tellen en bekijken van de chromosomen wordt ook als onvoldoende beschouwd voor een betrouwbare diagnose.
Daarom doet de dokter een genetische test. Deze methode wordt de FISH-test (fluorescentie in situ hybridisatie) genoemd. Bij deze procedure wordt wat bloed afgenomen bij de patiënt om de bloedcellen te onderzoeken op de afwezigheid van bepaalde componenten van chromosoom 17.
De diagnose van het Smith-Magenis-syndroom wordt als moeilijk beschouwd. Aangenomen wordt dat deze omstandigheid bijdraagt aan het lage aantal gevallen. Het is niet ongebruikelijk dat veel getroffen kinderen een andere diagnose krijgen, ook al hebben ze een sms. Dit zijn meestal ADHD (Attention Deficit / Hyperactivity Disorder) of autisme.
Het Smith-Magenis-syndroom wordt als ongeneeslijk beschouwd. Het verloop van de ziekte hangt af van de omvang, de leeftijd van diagnose en geschikte therapeutische maatregelen. Sommige patiënten kunnen meer dan 80 jaar oud worden.
Complicaties
Vanwege het Smith-Magenis-syndroom lijden patiënten aan verschillende mentale en fysieke aandoeningen en beperkingen. In veel gevallen worden familieleden of ouders ook getroffen door dit syndroom en hebben ze psychologische ondersteuning en behandeling nodig. De getroffenen lijden aan ernstige ontwikkelingsstoornissen en gedragsproblemen.
Ze hebben daarom speciale ondersteuning nodig en meestal ook hulp in hun dagelijkse leven. De drang tot zelfbeschadiging kan ook door ziekte worden gevoeld, zodat behandeling in een gesloten kliniek noodzakelijk kan zijn. Verder zijn er slaapproblemen of gehoorverlies. Het kleine postuur kan leiden tot pesten of plagen, vooral onder jongeren.
De getroffenen lijden ook aan visuele stoornissen en een hartafwijking. In ernstige gevallen kunnen ook epileptische aanvallen optreden, die tot de dood kunnen leiden. Vanwege de ernstige symptomen is de levensverwachting meestal aanzienlijk beperkt. De behandeling van het syndroom is puur symptomatisch. Helaas is het niet mogelijk om alle klachten volledig te beperken. Helaas kan het Smith-Magenis-syndroom ook niet worden voorkomen.
Wanneer moet je naar de dokter gaan?
Behandeling door een arts is altijd nodig bij het Smith-Magenis-syndroom. Omdat dit een erfelijke ziekte is, kan deze niet volledig worden genezen, zodat de getroffenen meestal levenslange therapie nodig hebben om de symptomen te verlichten. Indien de betrokkene kinderen wil, kan erfelijkheidsadvisering ook plaatsvinden om te voorkomen dat het Smith-Magenis syndroom wordt overgedragen op de nakomelingen.
Bij dit syndroom moet een arts worden geraadpleegd als de betrokkene lijdt aan ernstige ontwikkelingsstoornissen of ernstige gedragsproblemen vertoont. De getroffenen zijn erg onrustig en kunnen zich niet concentreren. In veel gevallen is zelfbeschadigend gedrag ook een aanwijzing voor het Smith-Magenis-syndroom en moet dit altijd door een arts worden beoordeeld. Bovendien vertonen de patiënten vaak oogklachten of hartafwijkingen. Bij deze klachten dient ook een arts te worden geraadpleegd.
Het Smith-Magenis-syndroom kan worden vastgesteld door een huisarts. Verdere behandeling wordt dan uitgevoerd door de betreffende specialist en hangt grotendeels af van de ernst van de symptomen.
Behandeling en therapie
Omdat het Smith-Magenis-syndroom wordt veroorzaakt door genetische factoren, kan het niet worden genezen. Om deze reden is de behandeling van de aandoening beperkt tot symptomen. Logopedie, ergotherapie en fysiotherapeutische maatregelen worden als nuttig beschouwd.
Communicatietraining met gebarentaal wordt ook als nuttig beschouwd. Ondanks alle therapiemaatregelen hebben de meeste patiënten hun hele leven hulp nodig. Bepaalde medicijnen kunnen ook worden gebruikt om de symptomen van het Smith-Magenis-syndroom te behandelen.
Dit kan sommige klachten verlichten. De toediening van het hormoon melatonine verbetert slaapstoornissen. Andere geneesmiddelen, zoals Risperidal, kunnen worden gebruikt om zelfbeschadiging tegen te gaan.
preventie
Smith-Magenis-syndroom is een genetische ziekte. Preventie is dus niet mogelijk.
U kunt dat zelf doen
De symptomen van de erfelijke ziekte verschijnen direct na de geboorte. Baby's kunnen natuurlijk geen adequate zelfhulpmaatregelen nemen.Bovendien zijn de gezondheidsstoornissen zo divers dat het voor de zieken niet mogelijk is om de situatie in voldoende mate te verbeteren.
Familieleden zijn daarom verplicht om de structuur van het dagelijks leven en de zorg voor de patiënt zo goed mogelijk voor de patiënt in te richten. Bij zelfdestructieve handelingen moet de betrokken persoon tegen zichzelf worden beschermd. Anders kunnen er ernstige complicaties en verslechtering van de gezondheid optreden. Omdat familieleden vaak overweldigd worden door de zorg voor de patiënt, is goede zorg nodig indien nodig. Tegelijkertijd is een stabiele sociale omgeving belangrijk, zodat de patiënt niet wordt blootgesteld aan extra stress.
Een groot aantal zieken heeft last van een verstoord slaap- en waakritme. Dit vormt een bijzondere uitdaging voor alle betrokkenen en ook het afwijkende gedrag is in het dagelijks leven niet altijd even gemakkelijk te hanteren. Zodra dierbaren hun emotionele en fysieke grenzen bereiken, moeten ze veranderingen aanbrengen. De organisatie van vrije tijd of het gebruik van psychotherapeutische ondersteuning kan helpen om een overeenkomstig evenwicht te ervaren. Omgaan met de ziekte is vaak zo stressvol dat anders psychologische complicaties kunnen optreden.
Nazorg
Nazorg voor het Smith-Magenis-syndroom (SMS) is in wezen gericht op het behandelen van de symptomen van de ziekte. Omdat sms genetisch is. Genezing is niet mogelijk. De reikwijdte van nazorgbehandelingen en onderzoeken kan bij SMS erg complex zijn.
Het spectrum van mogelijke symptomen varieert van lichamelijke afwijkingen en ontwikkelingsachterstanden (zowel lichamelijk als geestelijk) tot ernstige gedragsstoornissen, verstandelijke beperkingen en ernstige ziekten van de inwendige organen. De symptomen die door SMS worden veroorzaakt, genezen over het algemeen niet. De taak van de nazorg is het bereiken en behouden van een zo hoog mogelijke kwaliteit van leven voor mensen met VMS.
De focus van de nazorgbehandelingen ligt dan ook op het voortzetten van de therapeutische interventie. Als gevolg van de nazorg moeten kinderen en jongvolwassenen met SMS vooral geleerd hebben om zo goed mogelijk met de symptomen van de ziekte te leven. Naast levenslange medicamenteuze behandeling schrijven artsen ook kuren en logopedie, ergotherapie en muziektherapiebehandelingen voor als vervolgbehandelingen (regelmatig herhaald).
Als zelfhulpmaatregel kunnen nabestaanden tijdens de nazorg voor de sms-persoon een stabiele sociale omgeving creëren en het dagelijkse leven optimaal inrichten in het belang van de zieke. Als uw symptomen verergeren, heeft u mogelijk vervolgonderzoek nodig bij gespecialiseerde artsen.
.jpg)




.jpg)

.jpg)
.jpg)


.jpg)