Van de Middellange keten acyl CoA dehydrogenase-deficiëntie (MACD-deficiëntie) is een genetische stofwisselingsstoornis waarbij middellange vetzuren slechts onvoldoende worden afgebroken. Onder bepaalde omstandigheden kunnen gevaarlijke metabolische onevenwichtigheden optreden, die onder bepaalde omstandigheden fataal kunnen zijn. Als de therapie vroeg wordt gestart, kan de ziekte gemakkelijk onder controle worden gehouden.
Wat is middellange keten Acyl CoA-dehydrogenase-deficiëntie?

© royaltystockphoto - stock.adobe.com
Bij de Middellange keten acyl CoA dehydrogenase-deficiëntie de afbraak van middellange vetzuren wordt verstoord door genetische oorzaken. Deze kunnen niet meer voldoende worden gebruikt om energie op te wekken. Daarom wordt het lichaam gedwongen om koolhydraten en aminozuren in grotere mate af te breken.
Wanneer de energiebehoefte toeneemt door hogere fysieke stress, groeiprocessen of infecties, worden de koolhydraatreserves van het lichaam en de lichaamseigen eiwitten in grotere mate gebruikt voor de energieproductie. Hetzelfde geldt voor langere periodes zonder eten. Normaal gesproken wordt onder deze omstandigheden de afbraak van vetzuren geforceerd door bèta-oxidatie.
Dit is echter met één MACD-deficiëntie niet voldoende mogelijk. De resulterende verhoogde afbraak van glucose leidt tot gevaarlijke hypoglykemie. Door de geforceerde afbraak van aminozuren stijgt tegelijkertijd het ammoniakgehalte in het bloed. Carnitine, dat belangrijk is voor het energiemetabolisme, wordt verminderd omdat het verbindingen vormt met de zich opstapelende middellange vetzuren.
De verbindingen vertegenwoordigen tussenproducten van vetzuurafbraak Omdat de verdere afbraak van de middellange vetzuren wordt geblokkeerd door een defect enzym in het acyl-hydrogenase-complex, ontstaat een secundair carnitinedeficiëntie. Een grote afwijking van de corresponderende waarden kan tot levensbedreigende complicaties leiden. Indien onbehandeld, is ongeveer 25 procent van de metabole onevenwichtigheden die door deze aandoening worden veroorzaakt, dodelijk.
Over het algemeen is de middellange keten acyl-CoA dehydrogenase-deficiëntie een van de meest voorkomende stofwisselingsziekten en maakt deel uit van de onderzoeken als onderdeel van een pasgeboren screening. Een tijdige vaststelling van het verantwoordelijke genetisch defect is nodig voor een effectieve therapie. Deze ziekte kan gemakkelijk worden behandeld met geschikte therapeutische maatregelen.
oorzaken
De oorzaak van de middellange keten acyl-CoA dehydrogenase-deficiëntie wordt gevonden in een storing van het MCAD-enzym. Door een autosomaal recessieve mutatie van het MCAD-gen, dat zich op chromosoom 1 bevindt, zijn de vouweigenschappen van het enzym aangetast. Het resultaat is een verkeerd gevouwen enzym, dat wordt verwijderd en afgebroken als onderdeel van een eiwitkwaliteitscontrole.
Als resultaat is er niet genoeg acyl-CoA-dehydrogenase met middellange keten. Vetzuren met een middellange keten worden dan maar slecht afgebroken. Zoals eerder vermeld, is een autosomaal recessieve mutatie in het MCAD-gen verantwoordelijk voor deze ziekte. In het geval van autosomaal recessieve overerving heeft de aangedane patiënt twee defecte genen, waarvan één kopie is overgedragen van beide ouders.
Mensen met slechts één gemuteerd gen krijgen de ziekte niet. Daarom wordt de ziekte niet van generatie op generatie overgedragen. Als beide ouders elk heterozygoot zijn voor een defect gen, heeft het nageslacht een kans van 25 procent om MCAD-deficiëntie te ontwikkelen.
Symptomen, kwalen en tekenen
Acyl-CoA-dehydrogenasedeficiëntie met middellange keten wordt gekenmerkt door een neiging tot hypoglykemie, convulsies en comateuze toestanden. De eerste symptomen treden meestal op tussen de tweede levensmaand en het vierde levensjaar. Ze worden veroorzaakt door infecties of langdurige perioden van onthouding van eten en manifesteren zich als diarree, braken, verminderd bewustzijn en toevallen.
De lever kan vergroot zijn. Bij elke stofwisselingsstoornis worden de bloedsuikerwaarden verlaagd, de ammoniakwaarden in het bloed verhoogd en de carnitinespiegel verlaagd. Hoe groter de afwijkingen, hoe groter het risico om in een levensbedreigende coma te vallen. Er zijn echter ook vormen van MCAD-deficiëntie met veel mildere symptomen.
In sommige gevallen zijn er helemaal geen symptomen. Hier wijzen alleen laboratoriumtests op een overeenkomstig genetisch defect. Zelfs bij de meer ernstig zieke patiënten zijn er altijd symptoomvrije fasen tussen de metabole onevenwichtigheden.
Diagnose en ziekteverloop
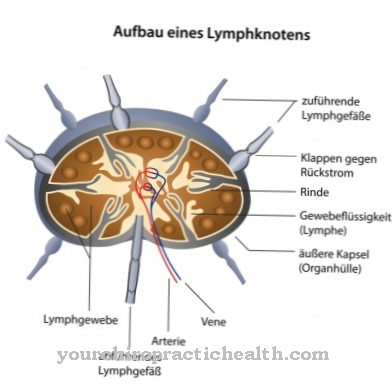
Een acyl-CoA-dehydrogenasedeficiëntie met middellange keten kan worden gedetecteerd in de context van een pasgeboren screening met behulp van tandem-massaspectroscopie. Bij deze methode worden fragmenten van acylcarnitines bepaald. De concentratie van octanoylcarnitine is hier de belangrijkste parameter. Als de niveaus verhoogd zijn, kan een DNA-analyse helpen om de diagnose te bevestigen.
In de acute fase van de ziekte kunnen hypoglykemie, verhoogde ammoniakgehaltes en verhoogde gehaltes van middellange vetzuren in de vorm van dicarbonzuren in de urine worden gedetecteerd. Er is ook hyperurikemie (verhoogde urinezuurspiegels in de urine) en tekenen van leverdisfunctie. Soms is het myoglobinegehalte in de urine ook verhoogd.
Complicaties
De middellange keten acyl-CoA dehydrogenase-deficiëntie kan in het ergste geval fataal zijn voor de patiënt. Om ernstige complicaties en gevolgschade te voorkomen, moet een vroege behandeling van deze ziekte plaatsvinden. In de meeste gevallen hebben de getroffenen last van ernstige krampen, die ook gepaard gaan met pijn.
Evenzo kan de middellange keten acyl CoA dehydrogenase-deficiëntie leiden tot coma of bewustzijnsverlies, zodat het dagelijkse leven van de persoon aanzienlijk wordt beperkt door de klacht. Bewustzijnsstoornissen treden op en de getroffenen lijden aan diarree en braken. In veel gevallen wordt de lever ook groter, wat ook gepaard gaat met pijn.
Als de betrokkene in coma ligt, kan deze aandoening ook een negatief effect hebben op de psyche van familieleden of ouders en kinderen en bij deze mensen tot depressie of andere psychische stoornissen leiden. Gewoonlijk kan acyl-CoA-dehydrogenasedeficiëntie met middellange ketens relatief goed worden behandeld als het vroeg wordt gediagnosticeerd.
Er zijn geen bijzondere complicaties. Pas bij een late diagnose en behandeling kunnen diverse complicaties optreden, waardoor de getroffen persoon bijvoorbeeld afhankelijk is van een operatie.
Wanneer moet je naar de dokter gaan?
Hunkeren naar voedsel en gerelateerde symptomen zoals misselijkheid en braken zijn tekenen van een gezondheidsprobleem. Bij concentratiestoornissen, malaise of afname van het prestatieniveau is een arts nodig. Bij pijn, krampen en een epileptische aandoening dient onmiddellijk een arts te worden geraadpleegd. Verlies van rijvaardigheid, verlies van eetlust of een verhoogde vatbaarheid voor infecties moeten worden onderzocht en behandeld. Aangezien de ziekte de eerste symptomen vertoont tussen de leeftijd van zes maanden en vier jaar, lopen met name kleine kinderen het risico de ziekte te ontwikkelen. Als de getroffen persoon een merkbare verandering in zijn gewicht vertoont, als er onverschilligheid of gedragsproblemen zijn, moet een arts worden geraadpleegd.
Verhoogde waakzaamheid is nodig in het geval van bewustzijnsstoornissen. Omdat het risico op een ongeval of letsel toeneemt door het verminderde bewustzijn van de betrokken persoon, is actie vereist. Als er een comateuze toestand optreedt of als er sprake is van bewustzijnsverlies, moet een ambulancedienst worden gewaarschuwd. Tegelijkertijd zijn eerstehulpmaatregelen vereist totdat deze arriveert. Zwelling van het bovenlichaam, vermoeidheid, uitputting alsook innerlijke rusteloosheid en verhoogde prikkelbaarheid dienen aan een arts te worden voorgelegd. De ziekte wordt gekenmerkt door de geleidelijke afname van alle symptomen. Ondanks de symptoomvrije periode is het raadzaam om een arts te raadplegen zodra er herhaaldelijk tekenen van een gezondheidsstoornis optreden.
Behandeling en therapie
Als de diagnose vroeg wordt gesteld, is de prognose voor de ziekte erg goed. Dan kunnen tijdig profylactische maatregelen worden genomen om metabole crises ook in moeilijke en stressvolle situaties te voorkomen. Het is belangrijk om langdurige onthouding van voedsel en infecties te vermijden.
Voedselverlof mag niet langer duren dan twee uur. Kennis van deze ziekte is ook noodzakelijk ter voorbereiding op eventuele operaties. Veel handelingen die niet absoluut noodzakelijk zijn, mogen daarom niet worden uitgevoerd. Als een operatie niet kan worden vermeden, is kunstmatige voeding met glucose-oplossing essentieel om metabole onbalans te voorkomen.
Zelfs een acute metabole onbalans kan alleen worden verholpen door een infusie met glucose-oplossing. Omdat er geen carnitine is, is orale toediening van carnitine ook zinvol. Daarnaast kan de toediening van riboflavine leiden tot een verbetering van de vetstofwisseling, omdat het een co-enzym is bij de afbraak van vetzuren.
Outlook & prognose
Bij aanwezigheid van een acyl-CoA-dehydrogenasedeficiëntie met middellange keten is de prognose positief als de diagnose zo vroeg mogelijk wordt gesteld. Onder bepaalde omstandigheden kan dit aangeboren metabolische defect echter leiden tot ernstige metabole onevenwichtigheden met fatale gevolgen.
Het probleem is dat een acyl-CoA-dehydrogenasedeficiëntie met middellange keten variabel is in het klinische beeld. Daarom kan een verkeerde diagnose niet worden uitgesloten. De symptomen van middellange keten acyl-CoA dehydrogenase-deficiëntie zijn al merkbaar bij pasgeborenen. Om de vooruitzichten voor de getroffenen te verbeteren, wordt metabolisme-screening tegenwoordig meestal op pasgeborenen uitgevoerd.
De levensomstandigheden zijn goed, zolang de betrokken persoon lange periodes van nuchter zijn of verhongering vermijdt. Hij zou een vetarm dieet moeten hebben dat voldoende hoeveelheden koolhydraten bevat. Daarnaast moet er voor een carnitinedeficiëntie gezorgd worden. Om een dreigende metabole onbalans te voorkomen, kan ook de toediening van glucose-elektrolyteninfusies worden overwogen.
Noodsituaties en dreigende metabole onevenwichtigheden vormen een hoog risico. Daarom moeten degenen die getroffen zijn door middellange-keten acyl-CoA-dehydrogenase-deficiëntie onmiddellijk doorverwezen worden naar een klinische behandeling. Degenen die getroffen zijn door MCAD hebben een nood-ID-kaart bij zich, die hen identificeert als patiënten met een middellange keten acyl-CoA dehydrogenase-deficiëntie.
De vooruitzichten voor mensen met middellange keten acyl-CoA dehydrogenase-deficiëntie zijn slechter als ze last hebben van infecties of als ze vasten. Nuchterheid voor een operatie brengt ook grote risico's met zich mee. Metabole onevenwichtigheden kunnen leiden tot hypoketotische hypoglykemie of metabole acidose. Als neurologische symptomen zoals hypotensie of lethargie optreden, kan bewusteloosheid of overlijden optreden.
preventie
Omdat middellange-keten acyl-CoA dehydrogenase-deficiëntie genetisch bepaald is, is er geen aanbeveling voor de preventie ervan. Als er meer dan één familielid is, is genetische counseling nuttig als er een kinderwens is. De defecte genen kunnen worden opgespoord door middel van DNA-onderzoek.
Als beide ouders een gemuteerd gen hebben, hebben de nakomelingen een risico van 25 procent op middellange-keten acyl-CoA-dehydrogenase-deficiëntie. Als bij de screening op pasgeborenen een MCAD-deficiëntie wordt gevonden, is een uitgebreide medische controle nodig om infectie te voorkomen. Het dieet moet worden aangepast aan een koolhydraatrijk en vetarm dieet.
Nazorg
Omdat peroxisomale ziekte is gebaseerd op een genetisch defect, kan therapie slechts enkele symptomen verlichten zonder de ziekte volledig te kunnen genezen. Om deze reden is er geen daadwerkelijke nazorgbehandeling in deze zin, maar een intensieve symptoombehandeling om sommige aandoeningen te verlichten.
Over het algemeen wordt de getroffenen en hun familieleden geadviseerd om een gezonde levensstijl te leiden met een uitgebalanceerd dieet om het immuunsysteem te ondersteunen. Hij zal ook bepaalde ontspannings- en mentale technieken voor stabilisatie voorstellen en adviseren over zoveel mogelijk vrijetijdsactiviteiten. Mentaal evenwicht maakt het gemakkelijker om met de ziekte om te gaan en kan soms het herstel bevorderen. Als de reeds getroffen ouders opnieuw kinderen willen krijgen, is een gedetailleerd genetisch onderzoek aan te bevelen om vooraf de kans op een ander ziek kind te bepalen.
U kunt dat zelf doen
Middellange keten acyl-CoA-dehydrogenase-deficiëntie is een stofwisselingsziekte die goed kan worden behandeld als deze vroeg wordt ontdekt. Een belangrijke zelfhulpmaatregel is het reguleren van uw stofwisseling door middel van voeding en voldoende beweging. Mensen die regelmatig medicijnen slikken of activiteiten ondernemen die de stofwisseling negatief beïnvloeden, dienen contact op te nemen met hun huisarts. Passende therapie kan het metabolisme optimaliseren en daarmee kan ook de MCADD goed behandeld worden.
Naast medische behandeling is ook alternatieve therapie mogelijk. Natuurlijke remedies zoals valeriaan of salie kunnen door hun pijnverlichtende en andere positieve effecten een goede invloed hebben op het genezingsproces. Om eventuele risico's en bijwerkingen te minimaliseren, dient het gebruik van dergelijke preparaten eerst met de verantwoordelijke arts te worden besproken.
Ouders van wie het kind aan MCADD is overleden, moeten traumatherapie zoeken. In het geval van een nieuwe zwangerschap dient screening in een vroeg stadium plaats te vinden, zodat een mogelijke middellange keten acyl-CoA dehydrogenase deficiëntie kan worden vastgesteld en wiegendood kan worden voorkomen. De getroffen kinderen hebben constante observatie nodig, zodat bij eventuele complicaties onmiddellijk contact kan worden opgenomen met de medische hulpdienst of de ambulancedienst.
.jpg)




.jpg)