Net zo Rothmund-Thomson-syndroom wordt een genetische huidziekte genoemd. Het wordt op een autosomaal recessieve manier overgeërfd.
Wat is het Rothmund-Thomson-syndroom?

© logo3in1 - stock.adobe.com
De Rothmund-Thomson-syndroom (RTS) is een van de genetische huidziekten. Vooral in het gezicht komt uitgesproken poikilodermie voor, die gepaard gaat met orthopedische en oogheelkundige klachten. Er is ook een verhoogd risico op het ontwikkelen van bepaalde kankers.
De naam Rothmund-Thomson-syndroom gaat terug op de beschrijving van twee artsen. Dit gebeurde voor het eerst in 1836 door de Duitse oogarts August von Rothmund (1830-1906). In 1936 publiceerde de Engelse arts Matthew Sydney Thomson (1894-1969) twee artikelen over erfelijke poikiloderma (Poikiloderma congenitale).
Deze ziekte is dezelfde als de ziekten beschreven door August von Rothemund. Om deze reden werd het later het Rothmund-Thomson-syndroom genoemd. Rothmund-Thomson-syndroom kan tot de zeldzame ziekten worden gerekend. Tot 2014 werden slechts 300 ziektegevallen geregistreerd. Het syndroom komt voor in gezinnen, wat typerend is voor erfelijke ziekten.
Vooral bloedverwanten of kleine gemeenschappen worden getroffen. Er is een evenwicht tussen de twee geslachten in termen van het aantal ziekten. Vanwege het lage aantal gevallen kan er echter geen precieze informatie worden gegeven. Bovendien wordt geen enkele etnische groep in het bijzonder getroffen door het Rothmund-Thomson-syndroom. De draaggolffrequentie van de mutatie is niet bekend.
oorzaken
In de geneeskunde is het Rothmund-Thomson-syndroom onderverdeeld in de subformulieren RTS-1 en RTS-2. Het is nog niet mogelijk geweest om duidelijk te maken wat de oorzaak van RTS-1 is. RTS-2 is terug te voeren op een heterozygote mutatie die is ontstaan in het RECQL4-gen. Het aangetaste gen is gecodeerd voor een helicase.
De meest voorkomende mutatie is de onzinmutatie. Bij tussen de 60 en 65 procent van alle patiënten zijn defecten in het RECQL4-gen de oorzaak van de ontwikkeling van het Rothmund-Thomson-syndroom. Beide typen RTS-1 en RTS-2 worden op autosomaal recessieve wijze overgeërfd.
Symptomen, kwalen en tekenen
Een typisch kenmerk van het Rothmund-Thomsons-syndroom is de onmiskenbare uitslag op het gezicht, die poikiloderma is. Deze uitslag is het belangrijkste symptoom van de erfelijke huidziekte Poikiloderma onderscheidt de ziekte ook van het RAPADILINO-syndroom.
Andere kenmerken van het Rothmund-Thomson-syndroom zijn dun hoofdhaar, het veelvuldig ontbreken van wimpers of wenkbrauwen, kleine gestalte, skeletafwijkingen, staar bij adolescenten en het optreden van een radiale clubhand. Bovendien verouderen de getroffen mensen voortijdig en ontwikkelen ze een aanleg voor kanker.
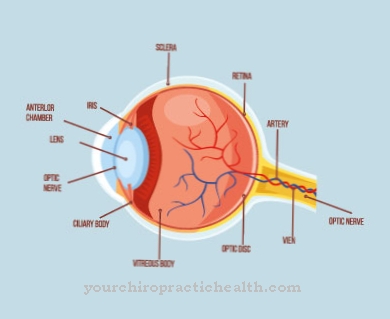
De symptomen variëren tussen RTS-1 en RTS-2. Bij RTS-1 treden poikilodermie, ectodermale dysplasie en juveniele cataracten van de ogen op. Patiënten met het subtype RTS-2 lijden ook aan poikilodermie. Daarnaast zijn er aangeboren misvormingen van de botten en een verhoogd risico op het optreden van osteosarcomen, kwaadaardige bottumoren tijdens de adolescentie. In verder leven kan het ook leiden tot huidkanker.
De misvormingen van het skelet bij het Rothmund-Thomson-syndroom zijn onder meer merkbaar aan een zadelneus, een radiale clubhand of een prominent voorhoofd. In sommige gevallen kunnen ze echter alleen met röntgenstraling worden gedetecteerd.
Diagnose en ziekteverloop
Er moet ook een onderscheid worden gemaakt tussen de twee vormen van het Rothmund-Thomson-syndroom in de diagnostiek. Er konden geen moleculair genetische triggers worden gevonden voor de ontwikkeling van RTS-1. Om deze reden wordt de diagnose gebaseerd op de aanwezige symptomen. Hiervoor is een speciale evaluatietafel gemaakt, die wordt gebruikt om de verschillende ziekteklachten te verzamelen.
Met behulp van puntwaarden kan de keuringsarts bepalen of RTS-1 aanwezig is. Als RTS-2 wordt vermoed, is het mogelijk om de mutatie in het RECQL4-gen te bepalen door een genetische test uit te voeren. Dit levert meestal ook het bewijs van RTS-2 op. Het Rothmund-Thomson-syndroom moet worden overwogen als patiënten osteosarcoom hebben. Er moet een differentiële diagnose worden gesteld voor het RAPADILINO-syndroom en het Baller-Gerold-syndroom, aangezien mutaties in het RECQL4-gen ook bij deze ziekten kunnen worden aangetroffen. Het Rothmund-Thomson-syndroom verloopt anders.
Ondanks hun zichtbare vroegtijdige verouderingsproces hebben de patiënten een relatief normale levensverwachting, mits er geen kwaadaardige kanker is. Sommige patiënten hebben een verhoogd risico op het ontwikkelen van secundaire maligniteiten. In deze gevallen hangt de prognose af van de kwaliteit van kankerscreening en kankerbehandeling. Als osteosarcomen optreden, ligt de levensverwachting van 5 jaar van patiënten tussen 60 en 70 procent.
Complicaties
Bij het Rothmund-Thomson-syndroom lijden mensen aan een aantal verschillende huidaandoeningen. Deze klachten hebben een zeer negatief effect op de esthetiek, waardoor veel patiënten zich ongemakkelijk voelen en last hebben van minderwaardigheidscomplexen of een verminderd zelfbeeld. Daardoor kunnen ook depressies of andere psychische klachten optreden. Rothmund-Thomson-syndroom kan leiden tot pesten of plagen, vooral bij kinderen.
Verder is er ook een kleine gestalte en verschillende misvormingen aan het skelet. De patiënten lijden aan een beperkte mobiliteit en in sommige gevallen aan de ontwikkeling van bottumoren. Huidkanker kan zich ook ontwikkelen als gevolg van de ziekte en kan de levensverwachting van de getroffen persoon aanzienlijk verkorten. Bovendien worden in veel gevallen de ouders of familieleden ook getroffen door de symptomen van het Rothmund-Thomson-syndroom en hebben ze ook last van psychische klachten.
Aangezien er geen oorzakelijke behandeling is voor het syndroom, kan alleen symptomatische behandeling worden uitgevoerd. Er zijn geen complicaties. De getroffenen zijn echter afhankelijk van stamceltransplantaties om de kankers te verslaan. In de regel kan een volledig positief verloop van de ziekte niet worden bereikt.
Wanneer moet je naar de dokter gaan?
Aangezien het Rothmund-Thomson-syndroom een erfelijke huidziekte is met een familiaire prevalentie, zijn contacten met een arts aan het begin van het leven onvermijdelijk. De poikilodermie in het gezichtsgebied die met de ziekte is geassocieerd, is echter niet het enige symptoom dat het leven moeilijk maakt voor de getroffenen.
Mensen met het Rothmund-Thomson-syndroom hebben ook last van orthopedische of oftalmologische complicaties. Mensen van beide geslachten hebben ook meer kans op kanker. De twee subtypes van het Rothmund-Thomson-syndroom komen echter relatief zelden voor. Er zijn niet meer dan 300-320 gevallen in de wereld gedocumenteerd.
Gezien de zeldzaamheid van het Rothmund-Thomson-syndroom, is het relatief moeilijk om een gespecialiseerde arts te vinden die het Rothmund-Thomson-syndroom kan diagnosticeren. Differentiatie van andere syndromen met vergelijkbare symptomen is noodzakelijk. Medische behandelingen zijn in de meeste gevallen onmisbaar, ook als er geen kans op genezing is. Rothmund-Thomson-syndroom kan alleen symptomatisch worden behandeld. Specialisten en specialisten uit verschillende disciplines streven naar een nauwe samenwerking.
De medewerking van dermatologen, orthopedisten, oogartsen, chirurgen of oncologen is de beste manier om de getroffenen te helpen een betere kwaliteit van leven te bereiken.
Therapie en behandeling
Er is momenteel geen therapie voor de oorzaak van het Rothmund-Thomson-syndroom. Om deze reden is de behandeling beperkt tot het verlichten van symptomen. Omdat de symptomen buitengewoon complex zijn, is het raadzaam om de patiënt interdisciplinair te laten behandelen. Dit betekent dat de therapie wordt uitgevoerd door artsen van verschillende specialismen. In het geval van het Rothmund-Thomson-syndroom omvat dit chirurgen, orthopedisten, oogartsen, oncologen en dermatologen (dermatologen).
Symptomatische therapie omvat een jaarlijks oftalmologisch onderzoek, behandeling van telangieectasieën en radiologische onderzoeken om osteosarcomen onder controle te houden. Stamceltransplantatie is een mogelijke nieuwe therapieoptie, die nog in klinische proeven is en tot dusver slechts bij twee patiënten is uitgevoerd. Eén patiënt onderging allogene beenmergtransplantatie. In het andere geval werd de behandeling uitgevoerd met stamcellen uit navelstrengbloed.
Uw medicatie vindt u hier
➔ Geneesmiddelen tegen roodheid van de huid en eczeempreventie
Helaas is het niet mogelijk om preventieve maatregelen te nemen tegen de uitbraak van het Rothmund-Thomson-syndroom. De aandoening is een van de erfelijke ziekten waarvan de oorzaak nog grotendeels onbekend is.
Nazorg
De vervolgzorg voor het Rothmund-Thomson-syndroom hangt af van het subtype en het beloop van de ziekte, evenals de gekozen behandelingsmaatregelen en de constitutie van de patiënt. In het algemeen zal de follow-up de verschillende symptomen bekijken en de volgende stappen bespreken. Chirurgische ingrepen vormen vaak een aanzienlijke belasting voor de patiënt.
Tijdens de follow-up worden de chirurgische littekens gecontroleerd om het succes van de behandeling te bepalen. Bovendien worden de patiënten aangemoedigd om te rusten. Behandeling met pijnstillers kan nodig zijn bij langdurige ziekten. Daarnaast hebben patiënten soms psychologische ondersteuning nodig. Onderdeel van de nazorg is ook de voorlichting over vorderingen in nieuwe therapiemethoden, zoals stamceltransplantatie of beenmergtransplantatie, die tot dusver met succes zijn uitgevoerd bij sommige patiënten.
Daarnaast omvat de follow-up een routineonderzoek van de verschillende fysieke functies die door de kanker zijn aangetast. Als er geen complicaties optreden, kan de patiënt worden ontslagen. Aangezien dit een uiterst zeldzame aandoening is die alleen symptomatisch kan worden behandeld, is nauwkeurige medische controle voor de patiënt raadzaam, zelfs na herstel. De vervolgzorg voor het Rothmund-Thomson-syndroom wordt uitgevoerd door een dermatoloog, orthopedist, oogarts, oncoloog of chirurg, afhankelijk van de symptomen.
U kunt dat zelf doen
Aangezien het Rothmund-Thomson-syndroom gepaard gaat met een verhoogd risico op kanker, moet de getroffen persoon zijn lichaam regelmatig onafhankelijk onderzoeken op onregelmatigheden of afwijkingen in de huid. Naast medische bezoeken voor medische onderzoeken, is het raadzaam om de hulp van andere mensen in het dagelijks leven in te roepen om gebieden die moeilijk toegankelijk zijn te verduidelijken voor mogelijke veranderingen in het uiterlijk van de huid.
Aangezien de erfelijke ziekte al in de eerste levensjaren afwijkingen vertoont, dient het opgroeiende kind in een vroeg stadium geïnformeerd te worden over de bestaande ziekte en het verdere verloop daarvan. Bij het dagelijks omgaan met de aandoening is het belangrijk om plotselinge situaties en onaangename verrassingen te vermijden. Daarom moet de nabije sociale omgeving ook worden geïnformeerd over de bestaande gezondheidsschade. Omdat er visuele veranderingen zijn, is het belangrijk om het zelfvertrouwen in het ontwikkelings- en groeiproces van het kind te versterken.
Omdat chirurgische ingrepen vaak worden gebruikt om de situatie te verbeteren, is mentale ondersteuning van het kind net zo belangrijk als het versterken van het immuunsysteem. Om een operatie het hoofd te bieden, heeft het organisme afweer nodig. Mentale kracht kan worden opgebouwd in samenwerking met een psychotherapeut. Bovendien boden de therapieën hulp om de uitdagingen van de ziekte emotioneel goed te kunnen verwerken. In sommige gevallen moeten ouders ook overwegen om psychotherapeutische behandeling te zoeken.
.jpg)
.jpg)
.jpg)






.jpg)

.jpg)
.jpg)
