De craniodiaphyseale dysplasie is een aangeboren skeletaandoening die gepaard gaat met hyperostose en sclerose van de aangezichtsschedel. De oorzaak is een genetische mutatie in de genen die de botstructuur remmen. De therapie is symptomatisch en richt zich op het stoppen van de ziekte.
Wat is craniodiaphyseale dysplasie?

© crevis - stock.adobe.com
Bij hyperostosen neemt de botstof op pathologische wijze toe. De hyperostose van de schedel is een groep ziekten die verband houdt met een dergelijke toename van botstof in het gebied van de schedel. Net zo craniodiaphyseale dysplasie wordt gekenmerkt door een aangeboren hyperostose van de schedel en is een skeletaandoening.
De Australische arts John Halliday beschreef de ziekte voor het eerst in het midden van de 20e eeuw. De frequentie wordt gegeven met een prevalentie van minder dan één geval op 1.000.000 mensen. Dit maakt skeletziekte een uiterst zeldzame dysplasie van de schedel.
Het complex van hyperostose en stenose van de gezichts- en schedelbeenderen is nu terug te voeren op een genetische oorzaak. Vanwege de weinige gedocumenteerde gevallen tot dusver, zijn niet alle verbanden tussen de ziekte definitief opgehelderd. Om deze reden zijn de therapiemogelijkheden momenteel ook beperkt.
oorzaken
In een groot aantal gevallen komt craniodiaphyseale dysplasie niet sporadisch voor, maar met familiale accumulatie. Zowel de autosomaal recessieve als de autosomaal dominante wijze van overerving zijn geïdentificeerd als de wijze van overerving voor de ziekte. De autosomaal dominante vorm van de ziekte is gebaseerd op een nieuwe mutatie in het SOST-gen. Het gen bevindt zich op locatie 17q21.31 en wordt beschouwd als een van de belangrijkste remmers van botvorming.
De mutatie van de SOST-genen is verantwoordelijk voor een groot aantal erfelijke botziekten, zoals VDB. Bij een mutatie kan het gen zijn remmende functies niet meer vervullen en breidt de botstructuur zich uit. Dit onderscheidt fundamenteel hyperostose van craniodiaphyseale dysplasie van andere hyperostosen.
De meeste van deze ziekten zijn gebaseerd op een disfunctie van de osteoclasten of osteoblasten. De genetische aanleg wordt geacht bewezen te zijn in verband met de ziekte. Welke andere factoren een rol spelen bij het ontstaan van de ziekte is niet definitief opgehelderd.
Symptomen, kwalen en tekenen
Het klinische beeld van craniodiaphyseale dysplasie wordt gekenmerkt door verschillende klinische criteria die al in de kindertijd tot uiting komen. Zieke baby's hebben meestal zwaar geblokkeerde neusholtes, waardoor ze ademhalingsproblemen kunnen krijgen. In het latere verloop van de ziekte is er in de meeste gevallen sprake van volledige obstructie van de neusholtes.
Vaak raken de traankanalen van de patiënt na dit fenomeen geblokkeerd. Op de onderkaak van de meeste getroffenen vormen zich geleidelijk groeiende neusuitstulpingen van benige substantie. De hyperostose van de gezichtsschedel vordert en ontwikkelt zich tot leontiasis ossea. In de meeste gevallen is de tandontwikkeling van de patiënt verstoord of vertraagd. De binnenkant van de schedel wordt smaller naarmate de ziekte voortschrijdt.
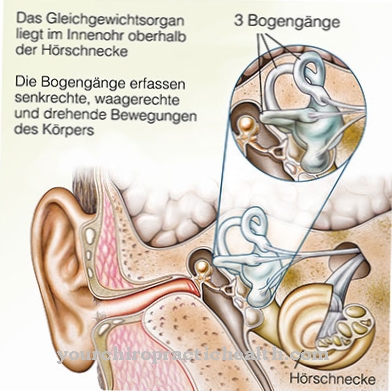
De vernauwingen hebben ook invloed op de foramina en veroorzaken opeenvolgende optische atrofie. Dit kan gepaard gaan met symptomen zoals gehoorverlies en meer of minder ernstige hoofdpijn. In sommige gevallen, omdat de binnenkant van de schedel steeds smaller wordt, krijgen de patiënten ook aanvallen. De schachten van de lange buisvormige botten worden steeds wijder.
Diagnose en ziekteverloop
De vroegst mogelijke diagnose en daaropvolgende therapie verbetert de prognose van patiënten met craniodiaphyseale dysplasie aanzienlijk. De arts vermoedt de hyperostose waarschijnlijk uit een visuele diagnose. Beeldvormingsprocedures worden beschouwd als het belangrijkste diagnostische hulpmiddel. Een röntgenfoto toont bijvoorbeeld extreme hyperostose en sclerose van alle schedelbeenderen.
De sleutelbeenderen of ribben kunnen verwijd lijken in de beeldvorming. De ontbrekende diafyses op de lange botten vallen duidelijk op. Een afgebakende, niet-verdikte cortex past ook in het klinische beeld. In termen van differentiële diagnose moet onderscheid worden gemaakt met ziekten zoals het Engelmann-syndroom. Moleculair genetische analyses zijn bijzonder geschikt voor een dergelijke differentiële diagnose. Het Engelmann-syndroom vertoont veranderingen in het TGFB1-gen bij mutatieanalyse, terwijl de craniodiaphyseale dysplasie het SOST-gen beïnvloedt.
Complicaties
Craniodiaphyseale dysplasie is een zeldzame, genetisch bepaalde skeletaandoening. Het symptoom manifesteert zich direct op de gezichtsschedel door een sterke toename van botstof met bijbehorende sclerose. De genetische mutatie is al zichtbaar in de kindertijd op basis van de vorm van de schedel en verkeerd geplaatste neusholtes, wat dreigende ademhalingsproblemen kan veroorzaken.
De resulterende gevolgen van craniodiaphyseale dysplasie brengen de getroffen patiënt vanaf de kindertijd talrijke levensbeperkende complicaties met zich mee. Als er geen tijdige klinische interventie is, zal de overmatige botgroei toenemen. Het binnenste van de schedel versmalt en de rijen tanden vormen niet voldoende. Het verdikkende botmateriaal vernauwt de gehoorgang en er is kans op gehoorbeschadiging en zelfs gehoorverlies.
Er is een toenemend gebrek aan ruimte in de schedelholte, en botafzettingen dringen de hersenen binnen. Ernstige hoofdpijn, toevallen, aangezichtsverlamming en epilepsie treden zowel op als een vermindering of regressie van de mentaal verworven vaardigheden. Ouders van wie de kinderen last hebben van craniodiaphyseale dysplasie, moeten daarom in een vroeg stadium klinische maatregelen zoeken.
Na het beeldvormend onderzoek vindt de differentiële diagnose plaats binnen de gegeven mogelijkheden. Er is momenteel geen basistherapie voor craniodiaphyseale dysplasie. Er worden pogingen gedaan om de ongecontroleerde progressie van botgroei en de gevolgen daarvan te beteugelen. Verschillende medicijnen en een calciumarm dieet vanaf de kindertijd zullen de patiënt helpen de symptomen te verminderen.
Wanneer moet je naar de dokter gaan?
Craniodiaphyseale dysplasie wordt vaak onmiddellijk na de geboorte vastgesteld. Als dit het geval is, zal de verantwoordelijke arts de ouders onmiddellijk op de hoogte stellen en daarna direct de behandeling starten. Als de dysplasie minder uitgesproken is, wordt de diagnose gesteld door de ouders. Een doktersbezoek is aangewezen als de pasgeborene ademhalingsproblemen of tranende ogen heeft. Externe afwijkingen, zoals de typische misvormingen aan het gezicht en de tanden, duiden ook op een ziekte die moet worden opgehelderd en behandeld.
Ouders die tekenen van gehoorverlies of epileptische aanvallen bij hun kind ervaren, moeten een arts raadplegen. Hetzelfde geldt als het kind vaak klaagt over hoofdpijn of de indruk wekt van hevige pijn. Tijdens de behandeling moet het kind regelmatig naar een arts worden gebracht. Dit zorgt ervoor dat het herstel zonder complicaties verloopt. Omdat craniodiaphyseale dysplasie gepaard gaat met een reeks symptomen, kan de therapie maanden of zelfs jaren duren. De huisarts raadpleegt hiervoor andere specialisten, altijd afhankelijk van de symptomen en klachten. Meestal zijn neurologen, internisten, oorspecialisten, chirurgen, fysiotherapeuten en psychologen bij de behandeling betrokken.
Behandeling en therapie
Een causale therapie voor patiënten met craniodiaphyseale dysplasie bestaat nog niet. Een dergelijke therapie kan in de toekomst mogelijk zijn via gentherapiebenaderingen. Op dit moment kan de ziekte echter alleen symptomatisch worden behandeld. Het belangrijkste doel van alle therapeutische maatregelen is om de overmatige botgroei te stoppen. Er zijn verschillende stappen te ondernemen.
Het voortschrijden van de ziekte kan bijvoorbeeld worden gestopt met medicatie. Calcitriol en calcitonine worden meestal als medicijnen gebruikt. Omdat de botstructuur afhankelijk is van calcium, kan een calciumarm dieet ook zinvol zijn. Dit specifieke dieet moet op de lange termijn worden gebruikt en idealiter het hele leven van de patiënt begeleiden.
De medicamenteuze behandeling van patiënten met het kunstmatige glucocorticoïde prednison heeft ook positieve effecten laten zien. Hoe vroeger de therapie wordt gestart, hoe veelbelovender het vooruitzicht. Met een extreem vroege behandeling kan hyperostose in de eerste levensjaren tot stilstand worden gebracht. Op deze manier worden de volgende symptomen drastisch verminderd.
Onder bepaalde omstandigheden kunnen ook chirurgische correcties worden aangebracht als onderdeel van de therapie. Dergelijke correcties hebben echter meestal weinig zin voordat het beloop van de ziekte onder controle is.
Outlook & prognose
Bij aangeboren maar zeer zeldzame craniodiaphyseale dysplasie is er een onherstelbare genetische mutatie. Daarom is de prognose voor de getroffenen niet erg goed. De medische professionals kunnen alleen proberen de symptomen en gevolgen van de toegenomen botgroei in het hoofdgebied te behandelen. Therapie kan het verloop van de ziekte alleen maar vertragen. Bij craniodiaphyseale dysplasie is de toename van botstof niet te stoppen.
Aangezien de huidige therapie-opties de onderliggende mutatie in de embryonale fase niet kunnen omkeren, zullen andere generaties van de getroffenen eraan lijden. Een familiale accumulatie is merkbaar bij craniodiaphyseale dysplasie. De symptomen die gepaard gaan met craniodiaphyseale dysplasie zijn al bij de zuigeling te zien. Omdat alle botadhesies plaatsvinden in het schedelgebied, worden de bovenste luchtwegen en het gehoor of het gezichtsvermogen erdoor beïnvloed.
Bovendien wordt het binnenste van de schedel in toenemende mate aangetast door botvorming. Dit beperkt de therapeutische benaderingen voor de volgende klachten. Hoe eerder de diagnose kan worden gesteld, hoe beter de prognose op lange termijn. Een calciumarm dieet remt de toenemende botgroei. Bovendien kunnen geschikte medicatie en prednison al in de kindertijd worden toegediend.
Met een interdisciplinaire behandelstrategie worden de beste resultaten behaald. Chirurgische ingreep bij craniodiaphyseale dysplasie heeft alleen zin als de progressie van de ziekte met succes is ingeperkt.
preventie
Tot dusver zijn er geen preventieve maatregelen voor craniodiaphyseale dysplasie. De ziekte is een genetische ziekte die verband houdt met een familiaire aanleg. Daarom kan alleen moleculair genetisch advies worden gebruikt als een soort preventieve maatregel.
Nazorg
In de meeste gevallen beschikt de getroffen persoon over zeer weinig vervolgmaatregelen. In sommige gevallen kan dit zelfs volledig worden beperkt, zodat de getroffen persoon afhankelijk is van een puur symptomatische behandeling van de ziekte. Zelfgenezing kan niet plaatsvinden omdat het een genetische ziekte is.
Daarom moet de betrokkene, als hij een kind wil, een genetisch onderzoek en advies laten uitvoeren, zodat de ziekte bij de kinderen niet terugkeert. De behandeling zelf wordt meestal uitgevoerd met behulp van verschillende medicijnen die de symptomen permanent kunnen verlichten en beperken. Het is altijd belangrijk om ervoor te zorgen dat het regelmatig wordt ingenomen, waarbij ook de juiste dosering moet worden aangehouden.
In het geval van kinderen dienen met name ouders te controleren of ze correct worden meegenomen en gebruikt. Regelmatige controles door een arts zijn ook nodig om de toestand van de ziekte permanent te controleren. De meeste misvormingen kunnen worden gecorrigeerd door chirurgische ingrepen. Veel van de getroffenen zijn ook in hun dagelijks leven afhankelijk van psychologische ondersteuning van hun eigen familie, wat een positief effect heeft op het verdere verloop van de ziekte. In de regel vermindert deze ziekte de levensverwachting van de patiënt niet.
U kunt dat zelf doen
In het geval van craniodiaphyseale dysplasie heeft de getroffen patiënt slechts beperkte effectieve maatregelen ter beschikking die een positief effect hebben op het beloop van de ziekte. Eerst en vooral is een passende therapie van de craniodiaphyseale dysplasie door een team van specialisten. De ziekte begint zich op jonge leeftijd te manifesteren, zodat het vooral de ouders zijn die bijdragen aan de kwaliteit van leven van de betreffende kinderen. Bij eventuele intramurale verblijven van de patiënten van het kind is het vaak logisch dat de ouders in het ziekenhuis aanwezig zijn en het kind daardoor emotionele steun krijgt.
In de loop van de ziekte treden vaak stoornissen op in de ontwikkeling van het gebit, waardoor de patiënten vaak afhankelijk zijn van orthodontische therapie. Ook bij het dragen van een beugel is uw eigen medewerking vereist. Er zijn ook aanwijzingen dat een calciumarm dieet de progressie van craniodiaphyseale dysplasie kan afremmen. Ook hier hebben de patiënten veel speelruimte wat betreft hun medewerking en daarmee hun kwaliteit van leven.
Vanwege de ademhalingsproblemen zien de patiënten af van bepaalde sporten, maar oefenen ze thuis ook krachtoefeningen bij een fysiotherapeut als dit medisch is toegestaan. Kinderen met craniodiaphyseale dysplasie krijgen voldoende onderwijs op speciale scholen.


.jpg)









.jpg)


.jpg)