De Allan-Herndon-Dudley-syndroom is een mutatie in het SLC16A2-gen dat de schildklierhormoontransporter MCT8 verandert en een verminderde opname van joodthyronine in spierweefsel en het centrale zenuwstelsel veroorzaakt. Door de mutatie hebben de getroffenen last van spierzwakte en vertragingen in de mobiele en mentale ontwikkeling. AHDS is ongeneeslijk en is tot dusver alleen behandeld met de toediening van trijoodthyroacetaat.
Wat is het Allan-Herndon-Dudley-syndroom?

Vertragingen in de fysieke, mentale of emotionele ontwikkeling van adolescenten en kinderen worden samengevat als ontwikkelingsachterstand of vertraging. Ontwikkelingsachterstanden kunnen verschillende oorzaken hebben. De trigger voor de vertraagde ontwikkeling kan bijvoorbeeld in het centrale zenuwstelsel liggen.
Dit is bijvoorbeeld het geval bij het Allan - Herndon - Dudley Syndrome (AHDS). Naast ernstige mentale retardatie wordt het syndroom gekenmerkt door stoornissen in de motorische ontwikkeling. De eerste beschrijving van het syndroom gaat terug tot 1944. Het ziektebeeld dat u beschrijft, is een erfelijke ziekte, d.w.z. een genetische aandoening.
De ziekte treft mannelijke zuigelingen in de meeste tot op heden gedocumenteerde gevallen. De ontwikkelingsstoornissen en hun gevolgen zijn in bijna alle gevallen vanaf de geboorte duidelijk zichtbaar. AHDS is een uiterst zeldzame ziekte.Om deze reden is het onderzoek naar het Allan-Herndon-Dudley-syndroom tot dusver vrij slecht.
oorzaken
AHDS is een erfelijke genetische aandoening die wordt veroorzaakt door een mutatie in het SLC16A2-gen. Dit is het coderende gen voor de zogenaamde schildklierhormoontransporter MCT8. Deze transporter bemiddelt bij de opname van joodthyronines in spier- en zenuwweefsel.
Door de mutatie treden er verstoringen op in de opname van schildklierhormonen, die het centrale zenuwstelsel verstoren en zo de ontwikkeling van de cellen van het zenuwstelsel belemmeren. Spierweefsel en hersenen raken verarmd door de mutatie-gerelateerde ontregeling van het actieve schildklierhormoon waarvan ze feitelijk afhankelijk zijn.
Het syndroom wordt doorgegeven als een X-gebonden recessieve overerving. Vrouwen kunnen de ziekte erven, maar vanwege hun dubbele X-chromosoomstructuur worden ze zelden ziek. Zieke mannen kunnen zich niet voortplanten.
Hoewel de mutatie genetisch is, spelen naast deze interne factor waarschijnlijk externe factoren een rol bij het ontstaan van de ziekte. Vanwege de zeldzaamheid en de beperkte onderzoeksbasis is de rol van deze externe factoren nog niet definitief opgehelderd.
Uw medicatie vindt u hier
➔ Geneesmiddelen tegen spierzwakteSymptomen, kwalen en tekenen
Allan-Herndon-Dudley-syndroom is een aangeboren ziekte die zich meestal manifesteert bij peuters of baby's. De getroffenen lijden aan meer of minder ernstige spierzwakte. Het spierweefsel van de kinderen is merkbaar onderontwikkeld. De zwakte van de spieren gaat al snel gepaard met gewrichtsmisvormingen.
Contracturen zijn ook vaak voorkomende symptomen. De mobiliteit van de kinderen wordt in toenemende mate belemmerd door contracturen en misvormingen. Om deze reden lijken de getroffenen vaak onnatuurlijk statisch of zelfs bewegingloos. Door het mutatiegerelateerde onderaanbod van schildklierhormonen hebben getroffenen vaak ook last van spierkrampen of maken onvrijwillige bewegingen met hun armen en benen.
Vaak kunnen de getroffenen zich niet zelfstandig verplaatsen. In de meeste gevallen worden de motorische stoornissen geassocieerd met ernstige psychische stoornissen. De meerderheid van de patiënten kan bijvoorbeeld niet praten. In individuele gevallen kan AHDS worden gekenmerkt door vele andere symptomen op het gebied van mentale en fysieke ontwikkeling.
Diagnose en verloop
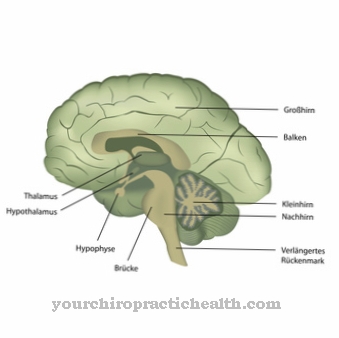
De arts vermoedt meestal eerst AHDS in de anamnese. In laboratoriumtests duidt een verhoogd T3-niveau met normale FT4- en TSH-waarden op het Allan-Herndon-Dudley-syndroom. Beeldvorming van het centrale zenuwstelsel is meestal onderdeel van de diagnose.
Spierzwakte als gevolg van motorneuronale aandoeningen kan worden uitgesloten van de differentiële diagnose. De prognose voor patiënten met het Allan-Herndon-Dudley-syndroom is relatief slecht. Tot dusverre is de ziekte ongeneeslijk. Studies hebben gesuggereerd dat de timing van de diagnose een cruciale rol kan spelen bij de prognose van de patiënt.
Complicaties
Zoals alle chromosonaal erfelijke aandoeningen, kan het Allan-Herndon-Dudley-syndroom niet curatief worden behandeld. De meest voorkomende bijwerking van Allan - Herndon - Dudley-syndroom - uitgesproken spierzwakte - kan worden behandeld met fysiotherapie. Een dergelijke behandeling gericht op het versterken van spieren kan pijnlijk zijn voor de patiënt.
Vooral kleine kinderen weigeren vaak therapie vanwege de pijn. Ondanks intensieve training leidt fysiotherapie niet altijd tot het gewenste succes. Het is vergelijkbaar met de logopedische ondersteuning van de Allan - Herndon - Dudley-patiënt. Hoewel de vermindering van de taalvaardigheid kan worden verbeterd met intensieve training, leidt de behandeling niet altijd tot succes vanwege de meestal hoogwaardige mentale beperking van de getroffenen.
Frustratie van de kant van de patiënt en een zware belasting voor het hele gezin is een van de ernstigste complicaties bij de behandeling van het Allan-Herndon-Dudley-syndroom. Spierkrampen en bewegingen van de extremiteiten die niet kunnen worden beïnvloed, kunnen worden behandeld met de toediening van spierverslappers. Complicaties zijn te zien in de soms ernstige bijwerkingen van de medicijnen.
Naast belasting van het maagdarmkanaal moeten vermoeidheid, een algemeen gevoel van uitputting en malaise worden genoemd. Langdurig gebruik van relanxantia beschadigt ook de lever en de nieren. Als het Allan-Herndon-Dudley-syndroom niet wordt behandeld, zullen de getroffenen geen significante vooruitgang kunnen boeken in termen van hun mentale of motorische vaardigheden.
Wanneer moet je naar de dokter gaan?
In veel gevallen is directe behandeling van het Allan-Herndon-Dudley-syndroom niet mogelijk. Om deze reden is de behandeling voornamelijk symptomatisch en gericht op de individuele klachten en vertragingen. In de regel moeten ouders dan een arts raadplegen als het kind spierzwakte heeft.
Dit kan merkbaar worden door uitputting of aanhoudende vermoeidheid. Medisch advies is ook nodig als het Allan-Herndon-Dudley-syndroom de mentale en motorische ontwikkeling vertraagt.
Als de behandeling niet plaatsvindt in de kindertijd, kan dit leiden tot aanzienlijk ongemak en beperkingen op volwassen leeftijd. Een arts moet worden geraadpleegd, vooral als de patiënt niet meer kan praten. Behandeling is ook nodig voor spierkrampen. Als het een acuut noodgeval is, kunt u rechtstreeks naar het ziekenhuis gaan of een ambulance bellen.
In de meeste gevallen zal het Allan-Herndon-Dudley-syndroom worden behandeld door een huisarts of door een kinderarts. De individuele klachten moeten echter door de betreffende specialist of therapeut worden onderzocht en behandeld.
Artsen & therapeuten bij u in de buurt
Behandeling en therapie
AHDS is een oorzakelijk onbehandelbare ziekte. Aangezien er geen therapieën beschikbaar zijn om de primaire oorzaak te verhelpen, is de ziekte tot dusverre niet te genezen. In de tussentijd suggereren de vorderingen op het gebied van gentherapie dat gentherapiebenaderingen binnenkort zullen worden goedgekeurd voor de dagelijkse klinische praktijk.
In hoeverre patiënten met het syndroom baat zouden hebben bij goedkeuring, is nog niet opgehelderd. Op dit moment is er ook geen gevestigde of gestandaardiseerde behandelingsoptie voor patiënten met AHDS op het gebied van symptomatische therapie. Enkele jaren geleden beschouwden onderzoekers de toediening van TRIAC als een mogelijke symptomatische therapieoptie.
TRIAC is een niet-klassiek schildklierhormoon, trijoodthyroacetaat. De toediening van het hormoon werd uitgevoerd in een klinische studie bij getroffen kinderen, maar kon geen zichtbare resultaten opleveren. De resultaten van het onderzoek zijn niet per se betekenisvol omdat de toediening van het hormoon relatief laat is begonnen.
Om deze reden werd TRIAC in 2014 nog als de best mogelijke therapie beschouwd. In één geval in 2014 werd tijdens de behandeling met TRIAC een significante verbetering van de motorische en mentale ontwikkeling vastgesteld. De therapie werd gestart bij de getroffen persoon in de vroege kinderjaren.
De resultaten van de onderzoeken tot nu toe geven dus aan dat het tijdstip waarop de therapie wordt gestart, een effect heeft op de therapieresultaten dat niet onderschat mag worden voor patiënten met AHDS. Begeleidende ondersteunende therapieën zoals ergotherapie en fysiotherapie of vroege interventie kunnen theoretisch worden gebruikt om de kwaliteit van leven en de vaardigheden van de patiënten te verbeteren. Er is echter nauwelijks bewijs voor de efficiëntie van een dergelijke benadering in verband met AHDS-patiënten.
Outlook & prognose
Het Allan-Herndon-Dudley-syndroom veroorzaakt bij de meeste patiënten een aantal verschillende symptomen. Eerst en vooral lijden de getroffenen aan ernstige spierzwakte. Dit betekent dat normale bezigheden of sporten niet meer gemakkelijk kunnen worden uitgevoerd voor de betrokkene. Er zijn ook grote vertragingen in de intellectuele en mobiele ontwikkeling. De concentratie van de patiënt is duidelijk beperkt en verminderd.
Er zijn nog steeds sterke krampen in de spieren en daardoor vaak onvrijwillige bewegingen of spiertrekkingen. Naarmate het Allan-Herndon-Dudley-syndroom vordert, kunnen de getroffenen niet langer praten. Het dagelijkse leven van de patiënt wordt aanzienlijk beperkt door het syndroom en de kwaliteit van leven wordt verminderd. In sommige gevallen zijn de patiënten dan in hun dagelijks leven afhankelijk van de hulp van andere mensen.
Het is meestal niet mogelijk om het Allan-Herndon-Dudley-syndroom causaal te behandelen. Om deze reden is de behandeling alleen symptomatisch. De getroffenen zijn afhankelijk van verschillende therapieën, die echter niet altijd tot een positief verloop van de ziekte leiden. In sommige gevallen beperkt het Allan-Herndon-Dudley-syndroom de levensverwachting van de getroffenen.
Uw medicatie vindt u hier
➔ Geneesmiddelen tegen spierzwaktepreventie
AHDS kan alleen worden voorkomen door genetische counseling. Mutatiedragers kunnen er bijvoorbeeld voor kiezen om geen eigen kinderen te hebben.
Nazorg
De behoefte aan nazorg voor het genetisch veroorzaakte Allan-Herndon-Dudley-syndroom treft alleen mannelijke baby's. Het probleem is dat er voor deze erfelijke aandoening geen geschikte therapie bestaat. De ernstige gevolgen van defecten aan transmitters van schildklierhormoon kunnen nauwelijks worden verbeterd. Pogingen om de getroffen kinderen verlichting te bieden door ze speciale schildklierhormonen te geven, zijn mislukt.
Het probleem is dat de basis van de ziekte meestal al in het lichaam van de moeder ligt. Ze schaden het ongeboren kind permanent. Vanuit dit oogpunt begint de behandeling te laat, namelijk pas na de bevalling. In de nazorg kan alleen de reeds aanwezige schade worden behandeld. Er is echter hoop. In 2014 werd een geval bekend waarin een baby met het Allan-Herndon-Dudley-syndroom met succes werd behandeld met TRIAC. Nazorg was nog steeds nodig omdat het kind niet te genezen was. Zijn symptomen werden tenminste verlicht.
Het Allan-Herndon-Dudley-syndroom wordt gedeeltelijk veroorzaakt door een defecte bloed-hersenbarrière. Dit is wat onderzoeken in het Cedars Sinai Hospital suggereren. De schildklierhormonen kunnen niet werken vanwege de defecte bloed-hersenbarrière. Dit geldt ook voor schildklierhormonen die worden toegediend nadat het kind is geboren. Biotechnologie of genetisch onderzoek kan mogelijk helpen. Alle pogingen tot behandeling mislukken momenteel. Dit heeft ook gevolgen voor de nazorg voor ernstig beschadigde kinderen.
U kunt dat zelf doen
Allan-Herndon-Dudley-syndroom is een ernstige aandoening die nog niet effectief is behandeld. Ouders kunnen echter nog steeds actie ondernemen om de therapie te ondersteunen.
Allereerst zijn regelmatige cognitieve training en fysieke activiteit belangrijk. Een uitgebreide therapie, die, afhankelijk van de ernst van het syndroom, kan bestaan uit spraak- en leestraining, maar ook uit algemene hersenoefeningen, biedt ook fysiotherapie-oefeningen. De training moet individueel worden afgestemd op de symptomen. De ouders van getroffen kinderen moeten er daarom voor zorgen dat de maatregelen optimaal worden gekozen en dat het kind niet overweldigd wordt.
In het geval van ernstige psychische stoornissen kan het kind permanente ondersteuning nodig hebben in het dagelijks leven. Een ambulante zorg kan een belangrijke verademing zijn voor de ouders. Een intramurale behandeling is net zo belangrijk, die thuis kan worden ondersteund door regelmatige controle van de symptomen.
Ouders moeten ook gebruik maken van psychologische begeleiding en, indien nodig, ook naar een zelfhulpgroep gaan, omdat contact met andere getroffen mensen het gemakkelijker maakt om met de ziekte om te gaan. Daarnaast krijgen ouders vaak belangrijke tips over hoe om te gaan met een ziek kind.
.jpg)












.jpg)

.jpg)