Net zo Syndroom van De Toni Debre Fanconi heet een genetische ziekte. Dit leidt tot een reabsorptie van verschillende stoffen in de nier.
Wat is het De Toni Debre Fanconi-syndroom?

© kras99 - stock.adobe.com
Het De Toni Debre Fanconi-syndroom is ook een van de namen De Toni Debre Fanconi Complex, Syndroom van De Toni Fanconi of Glucose Aminozuur Diabetes bekend. Er wordt bedoeld een nierresorptiestoornis van de proximale tubulus, die een deel van de niertubulus vormt. Syndroom van De Toni Debre Fanconi is een erfelijke ziekte en leidt tot reabsorptie van verschillende stoffen.
Deze omvatten aminozuren, glucose en anorganische fosfor. Dit leidt tot de pathologische uitscheiding van glucose, fosfaat en aminozuren. Het De Toni Debre Fanconi-syndroom werd voor het eerst beschreven door de Italiaanse arts Giovanni de Toni (1896-1973) en de Zwitser Guido Fanconi (1892-1979), die de ziekte ook zijn naam gaven.
Het Toni Debre Fanconi-syndroom is een van de zeldzamere ziekten. In Europa zijn ongeveer 50 gevallen van het infantiele type bekend. Secundaire vormen komen echter vaker voor.
oorzaken
Het Toni Debre Fanconi-syndroom kan worden onderverdeeld in drie verschillende vormen. Dit is het primaire idiopathische syndroom van Fanconi, dat op autosomaal recessieve wijze wordt overgeërfd, maar ook spontaan kan optreden. Het Fanconi-syndroom van het volwassen type verloopt minder ernstig dan het Fanconi-syndroom van het infantiele type.
De derde vorm is het secundaire Fanconi-syndroom. Dit komt voor bij erfelijke stofwisselingsziekten zoals cystinose of het syndroom van Lowe. Bovendien is het secundair aan het syndroom van Sjögren, amyloïdose, tumoren, vergiftiging of als een ongewenste bijwerking van medicijnen.
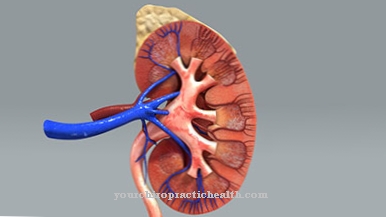
Het syndroom van De Toni Debre Fanconi is meestal een erfelijke ziekte. Hoewel het infantiele type van het syndroom wordt overgeërfd als een autosomaal recessieve eigenschap, is de overerving van het volwassen type autosomaal dominant. In het geval van het De Toni Debre Fanconi-syndroom is er een gegeneraliseerde resorptiestoornis van de proximale tubulus. Het pathomechanisme van het syndroom is nog niet volledig opgehelderd.
Aangenomen wordt dat er ischemie-gerelateerde ATP-deficiëntie of Na + - / K + ATPase-insufficiëntie is. Hierdoor worden verschillende stoffen zoals glucose, aminozuren of ionen zoals fosfaat niet meer uitgescheiden en uitgescheiden via de urine. Dit leidt op zijn beurt tot aminoacidurie, hypercalciurie waaronder hypokaliëmie, hyperfosfaturie, die gepaard gaat met een verstoring van de fosfaatbalans, en glucosurie inclusief osmotoïsche diurese.
Naast de erfelijke vorm van het De Toni Debre Fanconi-syndroom, die genetisch is, is er ook een verworven vorm. Dit wordt veroorzaakt door stofwisselingsziekten zoals fructose-intolerantie of de ziekte van Wilson, ischemie of nefrotoxische stoffen zoals zware metalen of medicijnen.
Symptomen, kwalen en tekenen
Symptomen van het De Toni Debre Fanconi-syndroom zijn afhankelijk van of het een infantiele of volwassen vorm van de ziekte is. Bij het idiopathische primaire syndroom dat optreedt in de kindertijd, treden symptomen op zoals een kleine gestalte, koortsaanvallen en braken tussen de leeftijd van 2 en 3 jaar. Bovendien lijden de zieke kinderen aan rachitis, wat vitamine D-resistent is.
Er is ook ernstige pijn in de botten. Zelfs pauzes zijn binnen het bereik van het mogelijke. Als het nierfalen, dat ook optreedt, niet wordt behandeld met bloedspoeling (dialyse) of een niertransplantatie, loopt het getroffen kind zelfs het risico op overlijden.
Als de symptomen van het De Toni Debre Fanconi-syndroom pas op de volwassen leeftijd verschijnen, zijn er normaal gesproken geen levensbedreigende gevolgen te vrezen. De volwassen vorm van het syndroom wordt merkbaar door spierhypotonie, polydipsie (pathologische dorst) of verzachting van de botten (osteomalacie).
Bovendien bestaat er een risico op complicaties door het tekort. Deze omvatten een lage bloedsuikerspiegel (hypoglykemie), spontane fracturen, neurologische aandoeningen en hypokaliëmische symptomen. Bovendien kan wereldwijd nierfalen optreden.
Diagnose en verloop
Om het syndroom van De Toni Debre Fanconi te diagnosticeren, kijkt de onderzoekende arts eerst naar de medische geschiedenis van de patiënt. Hij voert ook een lichamelijk onderzoek en een medisch laboratoriumonderzoek uit. Het is mogelijk om aminoacidurie of glucosurie te bepalen op basis van de urinestatus.
In het geval van multipel myeloom kan proteïnurie worden vastgesteld. Het fosfaatgehalte in het bloedserum wordt verlaagd. Af en toe kan hypokaliëmie ook worden vastgesteld. Er kunnen röntgenfoto's worden gemaakt om secundaire effecten van het De Toni Debre Fanconi-syndroom van osteomalacie of rachitis te diagnosticeren. Soms wordt ook een nierbiopsie (weefselverwijdering) uitgevoerd.
Het verloop van het De Toni Debre Fanconi-syndroom hangt af van de vorm. De prognose van de infantiele vorm wordt als ongunstig beschouwd, omdat zonder niertransplantatie de dood tussen tien en twaalf jaar optreedt als gevolg van nierinsufficiëntie. Als het daarentegen de volwassen vorm is, die alleen bij volwassenen wordt gezien, is de levensverwachting normaal.
Wanneer moet je naar de dokter gaan?
Als symptomen zoals een kleine gestalte, koortsaanvallen en braken worden opgemerkt tussen de leeftijd van twee en drie jaar, kan dit het De Toni-Debre-Fanconi-syndroom zijn. Een bezoek aan de kinderarts wordt aanbevolen als de symptomen niet vanzelf verdwijnen of als er aanvullende symptomen zijn. Over het algemeen moet u altijd een arts raadplegen als u tekenen van een ernstige ziekte heeft. Bij ernstige botpijn of zelfs breuken moeten de hulpdiensten worden gewaarschuwd of moet het getroffen kind naar het ziekenhuis worden gebracht.
Hetzelfde geldt voor tekenen van nierfalen of rachitis. Als de symptomen pas op volwassen leeftijd optreden, moet een arts worden geraadpleegd bij pathologische dorst, botproblemen en andere typische symptomen. Medisch advies is uiterlijk vereist bij complicaties zoals hypoglykemie, neurologische aandoeningen of spontane fracturen.
Naast de huisarts kan er ook een specialist in erfelijke ziekten of een internist worden ingeschakeld. Bij niet-specifieke symptomen neemt u best eerst contact op met de medische spoeddienst. Bij de genoemde klachten wordt in ieder geval medische opheldering aanbevolen.
Artsen & therapeuten bij u in de buurt
Behandeling en therapie
Bij de behandeling van het De Toni Debre Fanconi-syndroom is zowel causale als symptomatische therapie mogelijk. Als de patiënt lijdt aan de secundaire vorm, wordt de oorzaak van de primaire ziekte behandeld. Standaardtherapie omvat het toedienen van vitamine D3 of calcitriol om vitamine D-resistente rachitis te behandelen.
Hydrochloorthiazide wordt ook gegeven om het verlies van elektrolyten door de nieren te verminderen. Veel kleine maaltijden met veel eiwitten en koolhydraten zijn ook nuttig. Bovendien moet de patiënt dagelijks één tot drie liter vocht drinken. Tegelijkertijd is het belangrijk om de inname van keukenzout te verminderen. Daarnaast is het belangrijk om het verlies aan fosfaat, kalium en natrium te compenseren.
De acidose wordt op zijn beurt gecompenseerd door bufferoplossingen. Als er sprake is van een erfelijke proximale aandoening, is alleen symptomatische therapie mogelijk waarbij de patiënt natriumbicarbonaat, fosfaat, glucose en kalium krijgt. Regelmatige controle van de nieren en botten is ook belangrijk.
Outlook & prognose
Aangezien het syndroom van De Toni Debre Fanconi een genetische ziekte is, kan het niet causaal worden behandeld, maar alleen symptomatisch. Zelfgenezing komt niet voor bij dit syndroom.
Indien onbehandeld, kan het De Toni Debre Fanconi-syndroom leiden tot volledig nierfalen, wat uiteindelijk tot de dood leidt. De patiënten zijn dan afhankelijk van een donornier of dialyse.
Het syndroom kan ook leiden tot een kleine gestalte, braken of verschillende deficiëntieverschijnselen. De kwaliteit van leven en levensverwachting worden aanzienlijk verminderd. Bij kinderen leidt het syndroom ook tot een verstoorde en vertraagde ontwikkeling. Botbreuken en wondgenezing komen vaak voor.
Het syndroom van De Toni Debre Fanconi is meestal relatief goed te behandelen met behulp van medicatie. Dit verlicht alle symptomen en leidt tot een positief verloop van de ziekte. Patiënten zijn echter afhankelijk van levenslang gebruik van deze medicijnen, aangezien het syndroom niet volledig kan worden genezen. Bovendien moeten regelmatig onderzoeken van de inwendige organen worden uitgevoerd om complicaties te voorkomen. Met vroege therapie en succesvolle behandeling is er geen afname van de levensverwachting.
preventie
Het Toni Debre Fanconi-syndroom wordt gerekend tot de erfelijke ziekten. Er zijn dus geen preventieve maatregelen.
Nazorg
In het geval van het De Toni Debre Fanconi-syndroom heeft de getroffen persoon in de meeste gevallen maar een paar opties voor directe nazorg. De getroffen persoon moet in een vroeg stadium een arts raadplegen om verdere complicaties te voorkomen. Omdat het een genetische ziekte is, kan deze niet volledig worden genezen.
Als u kinderen wilt hebben, kan erfelijkheidsadvies en testen nuttig zijn om te voorkomen dat de ziekte terugkeert. Zelfgenezing komt niet voor bij het De Toni Debre Fanconi-syndroom. In de meeste gevallen hebben patiënten medicijnen nodig voor deze ziekte. Het is altijd belangrijk om het regelmatig met de juiste dosering in te nemen.
Bij kinderen moeten ouders de inname controleren. De getroffenen moeten ook veel drinken en indien mogelijk zout vermijden om de nieren niet onnodig te belasten. Aangezien het De Toni Debre Fanconi-syndroom tot ernstige nierproblemen kan leiden, zijn regelmatige controles en onderzoeken van de inwendige organen erg belangrijk. Vooral de botten en nieren moeten worden gecontroleerd. Het is niet universeel te voorspellen of het syndroom zal leiden tot een verminderde levensverwachting van de patiënt.
U kunt dat zelf doen
Naast de genetisch bepaalde erfelijke vorm van het De Toni Debre Fanconi-syndroom komt dit ook in een verworven vorm voor. De patiënt kan zelf geen maatregelen nemen tegen de erfelijke variant van de aandoening die een oorzakelijk effect hebben.
De infantiele vorm van de ziekte, die gewoonlijk merkbaar wordt tussen de leeftijd van twee en drie jaar, is bijzonder ernstig. Ouders moeten er dan op staan dat hun kind wordt behandeld door een arts die al ervaring heeft met deze vrij zeldzame ziekte. Specialisten zijn te vinden via de medische vereniging of met hulp van de zorgverzekeraar.
Bijkomende symptomen van de vaak voorkomende kleine gestalte, zoals spanning, spierpijn of beperkte motorische vaardigheden, kunnen meestal worden verlicht door vroegtijdig te starten met fysiotherapie. Zodra de kinderen emotioneel beginnen te lijden onder de kleine gestalte, moet ook een kinderpsycholoog worden ingeschakeld.
Als de ziekte is verworven en niet genetisch is, moet de eerste stap zijn om de oorzaak te achterhalen. Dit kan bijvoorbeeld een stofwisselingsziekte, fructose-intolerantie of vergiftiging met zware metalen zijn. Medicijnen kunnen ook verantwoordelijk zijn voor het verworven syndroom van De Toni Debre Fanconi.
Als de ziekte samenhangt met de levensstijl van de patiënt, bijvoorbeeld dieet of beroep, moet hij of zij zich aanpassen aan een verandering in levensstijl.Als een aanpassing van het dieet nodig is, moet een voedingsdeskundige worden geraadpleegd. Indien het beroep niet meer kan worden uitgeoefend, dient in een vroeg stadium de dienst voor loopbaanadvies van het uitzendbureau te worden ingewonnen. Ook in dergelijke gevallen adviseren de vakbonden kosteloos.

.jpg)



.jpg)




.jpg)

.jpg)



.jpg)

.jpg)